"Generic 3 mg revectina mastercard, bacteria 3 types smear."By: Carlos A Pardo-Villamizar, M.D.
 https://www.hopkinsmedicine.org/profiles/results/directory/profile/0008959/carlos-pardo-villamizar
Purchase cheapest revectina and revectinaMultilayered patella: similar radiographic findings in pseudoachondroplasia and recessive a number of epiphyseal dysplasia. Hemiepiphyseal stapling for angular deformity correction around the knee joint in youngsters with a number of epiphyseal dysplasia. Chondrodysplasia punctata: a boy with X-linked recessive chondrodysplasia punctata because of an inherited X-Y translocation with a present classification of these issues. Association of congenital deficiency of multiple vitamin K-dependent coagulation elements and the phenotype of the warfarin embryopathy: clues to the mechanism of teratogenicity of coumarin derivatives. Congenital rubella syndrome related to calcific epiphyseal stippling and peroxisomal dysfunction. Inherited chondrodysplasia punctata as a result of a deletion of the terminal quick arm of an X chromosome. Rhizomelic chondrodysplasia punctata and survival past one year: a evaluate of the literature and five case stories [see comments]. Dominant sex-linked inherited chondrodysplasia punctata: a distinct sort of chondrodysplasia punctata. Rhizomelic chondrodysplasia punctata: another peroxisomal disorder [letter to the editor]. Mesomelic dysplasia with punctate epiphyseal calcifications - a brand new entity of chondrodysplasia punctata? Clinical, radiological and biochemical classification of chondrodysplasia punctata. Dysplasia epiphysialis punctata: synonyms - stippled epiphyses, chondrodystrophia calcificans congenita (Hunermann). Metaphyseal dysotosis: description of an ultrastructural defect within the epiphyseal plate chondrocytes. Defective in-vitro colony formation of haematopoietic progenitors in sufferers with cartilage-hair hypoplasia and historical past of anaemia. Cartilage hair hypoplasia, metaphyseal chondrodysplasia type McKusick: description of seven patients and review of the literature. Fourier rework infrared microspectroscopic evaluation identifies alterations in mineral properties in bones from mice transgenic for sort X collagen. Of hedgehogs and hereditary bone tumors: re-examination of the pathogenesis of osteochondromas. Severity of disease and danger of malignant change in hereditary a number of exostoses. Tumor location affects the outcomes of easy excision for multiple osteochondromas in the forearm. Correlation of chronological age and bone age with the correction of ankle valgus by surface epiphysiodesis of the distal medial tibial physis. Natural historical past of multiple hereditary osteochondromatosis of the lower extremity and ankle. Abnormal scarring with keloid formation after osteochondroma excision in youngsters with multiple hereditary exostoses. Dyschondrosteosis, a hereditable bone dysplasia with attribute roentgenographic options. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is important for osteoblast differentiation and bone growth. Localization of a gene for autosomal dominant Larsen syndrome to chromosome area 3p21. Mutations within the gene encoding filamin B disrupt vertebral segmentation, joint formation and skeletogenesis. Congenital knee dislocation in a patient with Larsen syndrome and a novel filamin B mutation. Goldberg Syndromes of Orthopaedic Importance the word syndrome is derived from a Greek word which means to run together. When several relatively unusual anomalies happen in the same individual, it may be nothing greater than coincidence. However, if all of the anomalies result from the identical cause, or occur in the same pattern in other kids, that exact combination of birth defects known as a syndrome. In such cases, appropriate referrals must be made to a geneticist to help in syndrome identification, order acceptable confirmatory checks, and organize for management of the nonorthopaedic manifestations of the syndrome. The analysis of a child for a syndrome features a household historical past, a systems review, and a search for minor dysmorphic options, such as irregular palm creases or abnormal shape of digits or toes. During fetal improvement, cell signaling pathways are activated in a coordinated method to permit cells to divide, differentiate, move, and die off, in the end resulting in a normally fashioned particular person. Such pathways could be dysregulated by a mutation in a key pathway member, by fetal environmental factors. Even within a household during which all of the members carry the identical causative gene mutation, some people are minimally affected, whereas others have all the findings of the syndrome. This could also be due to the presence of modifying genes, which is probably not inherited in the same means because the gene mutation that causes the syndrome, or as a outcome of fetal environmental elements that modify the style in which the pathways are activated. In addition, totally different mutations in the identical gene can cause completely different syndromes, as a end result of the merchandise of various mutations have completely different mobile functions. Such is the case with the dystrophin gene, which causes each Duchenne and Becker muscular dystrophies. Information concerning the etiology of a syndrome is necessary, as a end result of it has implications for the parents as to the chance of recurrence in subsequent pregnancies, and should maintain the key to the event of novel therapies. The rapid tempo of primary analysis in developmental biology and genetics makes it difficult for a standard textbook to present the most upto-date details about syndrome etiology. Discussions in regards to the threat of subsequent pregnancies are in the realm of the genetic counselor. The significance of understanding syndromes is in recognizing associated medical abnormalities that may be life threatening, adversely affect orthopaedic outcomes, or may influence surgical timing and management. For instance, within the case of Marfan syndrome, starting a child on beta-blockers can prevent a catastrophic cardiovascular occasion. Nomenclature can confuse syndrome identification, as a end result of a single syndrome may have a number of names. Many syndromes are brought on by a mutation in a gene, and the causative gene has been identified in most such syndromes. Furthermore, gene names are regularly unrelated to clinical findings associated with a given syndrome. An ideal nomenclature, which would give details about clinical findings and etiology, has yet to be developed. This information is used for narrowing down the diagnosis to only a handful of syndromes. In many instances, the ultimate analysis may be made on the medical and radiographic findings alone [e.
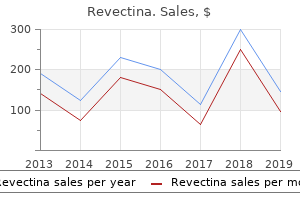
Discount 3mg revectina otcThey referred to as their pediatrician who evaluated him and referred him for a possible an infection in the proper knee. The previous history reveals that 2 weeks in the past he had a fever and cough that lasted for 5 days. The clinician understands that though the historical past is typical for a patient with transient synovitis of the hip, it might also be in preserving with septic arthritis, osteomyelitis, or LeggCalv鮐erthes disease. A 4-year-old boy will typically describe exactly where it hurts and should level to the groin, thigh, or the knee. It is important to do not neglect that pain may be referred, and a hip problem presenting as knee ache is a classic instance of referred pain. The clinician asks if the pain is fixed or intermittent as children with infections are inclined to have constant pain. The fever and cough 2 weeks ago is an important a part of the historical past, as an higher respiratory an infection is often a precursor of transient synovitis of the hip. A patient with acute septic arthritis involving the hip would typically have a history of 1 to 2 days of severe ache, whereas a affected person with juvenile arthritis could have had intermittent low-grade ache for several weeks to months. The bodily examination can start by asking if the affected person will stand and take a few steps. If the kid has an antalgic (painful) gait on the proper facet, the stance part of gait would usually be shortened and he would lean over the painful hip. When a baby stands on one leg, the hip abductor muscles contract to maintain the pelvis up on the opposite facet and keep away from falling. The mixture of weight-bearing and contracted abductor muscular tissues markedly increases the joint reactive forces in the hip. Since the child is having pain, the uninvolved aspect is examined first earlier than continuing to the symptomatic aspect. The hips are examined with the kid in the supine position, and the clinician looks for any asymmetry as the hips are attempted to be taken by way of a full range of motion. The vary of motion of both hips, especially inner rotation, ought to be symmetric. This is especially noted in attempting to internally and externally rotate the hip, with the hip and knee in 90 degrees of flexion. This is more snug for the affected person, and if he has severe ache with log rolling, the ache might be unbearable with the hip and knee in flexion. In this place, if the knee is pushed toward the inspecting table, it transmits a tensile force to the sacroiliac joint. This boy has transient synovitis of the best hip, so exercise modification and shut follow-up are really helpful to make sure that the symptoms are resolving. The clinician palpates the iliac crests while the affected person stands on the left lower extremity. In single limb help on the left decrease extremity, the best iliac crest should rise as the left hip abductor muscle tissue contract to help the pelvis. The Trendelenburg check can also be optimistic if the affected person has an irritable left hip. The downside was first famous by the maternal grandmother when she started strolling at 13 months of age. The household is concerned that the issue is getting worse they usually wish to know if she should have arch supports or particular footwear. The previous medical history in addition to the delivery and developmental history are inside regular limits. The family history reveals that her father had flat feet and wore special sneakers until he was 2 years old. The clinician critiques the main points of the history to determine when the flat ft have been first famous. In distinction, a rigid pes planus deformity, similar to that seen in a baby with a tarsal coalition, is usually not noted until the child is 10 years of age, when the cartilaginous bar begins to ossify causing pain and decreased movement of the foot. The clinician can gently "log roll" the extremity with the hip and knee in full extension to determine the severity of the pain. This is extra snug for the patient and if he has extreme pain with log rolling, the ache might be unbearable with the hip and knee in flexion. Ligamentous laxity may be detected by asking the patient to try to hyperextend the little finger metacarpophalangeal joint (>90 degrees indicates laxity). A patient with a planovalgus deformity will typically toe-out, whereas a affected person with a cavovarus deformity will often toe-in. Before focusing on the ft, a general physical examination of the again, upper, and lower extremities is performed. A patient with versatile pes planovalgus will often have generalized ligamentous laxity. Flexible pes planus is widespread and is most likely brought on by excessive laxity of the ligaments and joint capsules, allowing the tarsal arch to collapse with weight bearing. The knee is gently pushed towards the examination desk (arrow) transmitting a tensile force to the sacroiliac joint that may trigger ache if the sacroiliac joint is infected. Ligamentous laxity could be detected by asking the affected person to try and touch the thumb to the volar surface of the forearm. The arch returns when the kid is sitting, as a end result of the weight-bearing drive that caused the collapsed arch is relieved. The household first famous that he was limping on the proper aspect after a soccer recreation 2 months ago. The limp went away, however three days later he complained of right groin ache and they noticed that he was limping again. They went to their pediatrician, who questioned whether he could have pulled a muscle while enjoying soccer. The pediatrician documented that he was within the 5th percentile for peak and the ninety fifth percentile for weight. It is unusual to sustain a groin muscle harm at this age, and signs from a groin muscle harm will sometimes enhance in 2 to three weeks. If the child was black, the clinician could contemplate that he may have sickle cell disease with a bone infarct involving the proximal femur. A bone infarct in a patient with sickle cell disease will sometimes present with the sudden onset of ache within the groin, somewhat than having pain and limping for 2 months. A child with Legg-Calv鮐erthes illness will often complain of pain within the groin and may have short stature and a delayed bone age. A: In standing, a affected person with a versatile pes planus has a collapsed medial longitudinal arch (arrow), a valgus hindfoot, a supinated forefoot, and an externally rotated forefoot. B: If the pes planus is flexible, the arch will correct when she stands on her tip toes (arrow). When the good toe is dorsiflexed on the metatarsophalangeal, the arch is restored (arrow) because of the windlass impact of the plantar fascia. Children with Legg-Calv鮐erthes disease undergo several stages that could be summarized as damaging and reparative phases (19). A child with multiple epiphyseal dysplasia could have quick stature and may have ache and a limp. Multiple epiphyseal dysplasia is inherited as an autosomal dominant trait, so there could additionally be a family historical past of the dysfunction.
Generic 3 mg revectina mastercardAcute liver failure (fulminant hepatitis) Establishing the analysis of Wilson illness in the patient with acute liver failure could also be particularly troublesome. Liver biopsy may not be potential due to coagulopathy; renal failure could preclude collection of urine; ceruloplasmin may be low due to the liver failure itself; and K-F rings are often absent due to the younger age of the sufferers. This presentation may appear just like acute fulminant viral, autoimmune, or toxin induced hepatitis, although several clues should counsel Wilson disease. A Coombs-negative hemolytic anemia, with elevated reticulocyte depend, brought on by rapid launch of massive quantities of copper from the necrosing liver is attribute of Wilson disease, causes an especially high serum bilirubin (reaching 600 mg/dL), and makes this the more than likely analysis [1]. Occasionally hepatitis A virus triggers hemolysis in sufferers with glucose-6-phosphate dehydrogenase deficiency or thalassemia trait; however, hepatitis A virus infection can shortly be excluded serologically. Other findings that ought to suggest Wilson disease embody comparatively low serum aminotransferase for fulminant hepatic failure of 2ͱ0� upper limit of regular and an abnormally low serum alkaline phosphatase degree for affected person age. Results of genotyping are unlikely to be available when selections surrounding liver transplantation need to be made urgently. Transjugular liver biopsy can permit tissue to be obtained even when a big coagulopathy is current offered the affected person is secure enough to be moved to the radiology suite. Characteristic histologic findings of Wilson illness, corresponding to fatty change, periportal glycogenated nuclei, and an elevated hepatic copper content, aid in the prognosis. If the analysis of Wilson illness escapes detection, all patients with acute liver failure will die of hepatic or renal failure. Even when appropriately diagnosed, these sufferers nearly by no means get well despite copper chelation therapy, plasmapheresis, or postdilution hemofiltration and require urgent liver transplantation. In many patients, it will be obvious that liver transplantation will be required whatever the underlying analysis. A excessive diploma of suspicion is critical to pursue slitlamp examination for a K-F ring, investigate for a optimistic household history, or acknowledge neuropsychiatric symptoms. For this cause, all children age 3ʹ years or older with cirrhosis should endure evaluation for Wilson illness as described. Yes All regular No Liver biopsy Homozygote Normal or heterozygote No Wilson disease Normal histology, liver Cu2+ < 100 ֧/g dry weight Genetic Counseling Liver Cu2+ >250 ֧/g dry weight Any of following: signs indicators of liver dz. Attributing elevated aminotransferases as being caused by non-alcoholic fatty liver disease in overweight or obese patients has the potential of overlooking Wilson illness in a significant variety of patients. Institution of copper chelation at this stage of Wilson illness assuredly will forestall critical hepatic or neurologic sequelae if cirrhosis has not yet developed. Asymptomatic sufferers with KayserΆleischer rings In an asymptomatic patient for whom a K-F ring is found on routine ophthalmologic examination, plasma ceruloplasmin should be assessed. Elevated urine copper excretion, low plasma ceruloplasmin, and a K-F ring establish the analysis. However, if liver or spleen is enlarged, or liver blood checks are abnormal, a liver biopsy should be thought-about. Asymptomatic siblings of sufferers with Wilson illness Asymptomatic siblings and different first-degree family members of a Wilson illness affected person must be screened for the disease after 2 years of age, except hepatomegaly or irregular aminotransferase ranges are found earlier. If all checks are regular, it is very unlikely that the relative has Wilson disease. However, if any of these tests are irregular, a liver biopsy must be performed for histology, electron microscopy, and quantitative copper analysis. Finding two disease-causing mutations or informative microsatellite markers will set up the analysis of Wilson disease and preclude the need for liver biopsy, unless there are signs or symptoms of chronic liver disease. The hallmark of medical administration in Wilson illness is lowering or chelating the stored copper and stopping copper from reaccumulating. This is completed by instituting remedy with certainly one of a number of copper-chelating agents, a low copper food plan, oral zinc, and, possibly, antioxidants (Table 28. Stringent dietary restriction of copper-containing foods is impractical, though it is suggested that sufferers keep away from foods with very excessive copper content, corresponding to chocolate, nuts, legumes, mushrooms, shellfish, and liver. British AntiLewisite was then launched and shown to effectively chelate copper but the want for daily intramuscular injections limited its usefulness. These are D-penicillamine (,-dimethylcysteine), trientine, zinc, and the investigational drug ammonium tetrathiomolybdate. There is only one recently revealed randomized trial evaluating two of these therapies [48]. Published information embody primarily single center expertise and mixed heart reviews. The most printed expertise is with D-penicillamine as a end result of it was the primary orally effective drug developed for the treatment of Wilson disease. D-Penicillamine the paramount work of Walshe clearly demonstrated the advantages of D-penicillamine in Wilson illness [18]. The pharmaceutical type is D -penicillamine (referrred to as D-penicillamine or penicillamine), as L -penicillamine is toxic. The major motion of penicillamine was initially thought to be a "decoppering effect," though there are conflicting knowledge as to whether it actually reduces whole copper content material of the liver and different organs. Alternatively, a "detoxing" effect has been proposed in which copper is immediately complexed to the drug and induction of metallothionein synthesis occurs. It is beneficial to begin penicillamine therapy with 250͵00 mg/day and enhance by 250 mg increments every four 7 days to a maximum dose of 1000ͱ500 mg/day in two to 4 divided doses in an grownup, given ideally no much less than 30 minutes earlier than or 2 or more hours after meals as meals inhibits its absorption [1]. In children, the goal dose is 20 mg/kg day by day, rounded to the closest multiple of 250 mg and given ideally in three divided doses. After stabilization, the dose can be divided into two or three every day doses not to be given with meals. Penicillamine might have an antipyroxidine effect; subsequently, all handled sufferers should also receive 25͵0 mg pyridoxine day by day [1]. During the primary month of penicillamine therapy, the affected person must be monitored weekly for fever or rash. A full blood depend and platelet rely, urinalysis, renal and liver blood tests must be obtained every 1 to 2 weeks. If the patient responds appropriately with decision of symptoms and normalization of liver blood tests, monitoring is carried out every 1 to 3 months for the first yr, and every 6 to 12 months thereafter. Abnormalities in liver blood tests could persist for a minimum of a year of treatment; nevertheless, the pattern should be toward enchancment within the first 6 months of remedy. An annual 24-hour urinary copper excretion is useful in monitoring persistent penicillamine remedy. Initially, as a lot as a number of grams of copper could also be excreted in 24 hours; nevertheless, after months to years of chelation, as little as 200 to 500 g of copper ought to be excreted per day [1]. If urinary copper excretion is <200 g/24 h, poor adherence with chelation therapy must be suspected or overtreatment and excess copper elimination. In those with non-adherence, non-ceruloplasmin-bound copper is elevated (>15 g/dL) and in these with overtreatment, values are low (<5 g/dL).
Buy generic revectina 3 mg onlineThis is an entire or partial double radiodensity, which is never seen in other conditions. Upper extremity involvement is much less severe; there could additionally be irregularities within the proximal and distal humerus and radius. The humeral head involvement in adulthood has been termed a "hatchet-head" look, and outcomes from undergrowth of the pinnacle and neck. The spine may be regular, or it could have slight endplate irregularities or ossification defects on the anterior margins of the vertebrae (178). However, with substantial delay in epiphyseal ossification, recurrent deformity has been reported (179). Pain is more likely to occur in instances by which avascular necrosis has supervened (172). Acetabular shelf augmentation is a worthwhile process in these cases (180, 181). Severe deformities could also be corrected by osteotomy at maturity within the femur or tibia, depending on the positioning of abnormality. Patients having a double-layered patella could have symptoms because of the relative movement of 1 over the opposite. This state of affairs could also be treated by excision of one or fusion of the two segments, as appropriate (183). It results not so much from malalignment of the joints, but from intrinsic defect in cartilage. It produces stiffness, from an early age, and ache resulting in a total joint arthroplasty. Differences from normal osteotomy embrace a shallow or an anteverted acetabulum or slender femoral canal (184). Even the shoulder is commonly affected by degeneration, and shoulder arthroplasty may be essential (185). Skeletal findings within the extremities embody limb-length inequality, coxa vara, and clubfoot or different foot deformities (206). Spinal findings embody atlantoaxial instability, congenital scoliosis, spinal stenosis or kyphosis (196, 207, 208). The look is of small flecks of calcium, "which appear as if paint had been flecked on by a brush" (210). This skeletal dysplasia is named for a radiographic finding of calcification in skeletal cartilage, which can come up through several different pathways (186). There are many totally different types, the most typical being an X-linked dominant kind (Conradi-H𮥲mann syndrome), followed by an autosomal recessive rhizomelic sort (which is often fatal in infancy) and a uncommon X-linked recessive kind. There are numerous conditions that may present with the identical phenomenon: Zellweger (cerebrohepatorenal) syndrome, gangliosidosis, rubella, trisomy 18 or 21, vitamin K deficiency, hypothyroidism, or fetal alcohol or hydantoin syndromes (188ͱ93). The genetic defect and pathogenesis of the Conradi-H𮥲mann syndrome is related to a defect in sterol metabolism (186). The ultimate frequent pathway of the varied types is a defect in cholesterol synthesis (196) or peroxisomal enzymes. Histologic examination exhibits perilacunar calcifications all through the cartilage matrix (197). Patients with Conradi-H𮥲mann syndrome are characterized by hypertelorism, a depressed nasal bridge, and a bifid nasal tip (198Ͳ01). In addition, many have alopecia, congenital heart and/or renal malformations, and mental retardation. In rhizomelic chondrodysplasia punctata, findings include microcephaly, a high incidence of congenital cataracts, progress retardation, and a well-formed nasal bridge (202Ͳ05). Some have feeding difficulties, and many succumb to dying from respiratory points or seizures in the first yr. Diagnosis may be made by amniocentesis, with measurement of plasmalogen biosynthesis and phytanic acid oxidation. A: Diffuse punctate epiphyseal calcifications in infancy are a trademark for which chondrodysplasia punctata was named. Because of the chance of cervical instability or stenosis, each patient should have a lateral cervical radiograph and, if instability appears potential, a flexion-extension view. It could require early fusion if progression is documented and the affected person, medically, is a candidate. Anterior structural grafting, adopted by posterior fusion and solid immobilization, has the highest price of success (196). Coxa vara should be handled whether it is symptomatic and the neck-shaft angle is <100 levels. Metaphyseal chondrodysplasias are a family of issues leading to metaphyseal irregularities and limb deformity (211Ͳ14); however, the epiphyses are uninvolved. There are 4 named issues in this household: McKusick kind, Schmidt type, Jansen metaphyseal dysplasia, and Kozlowski kind. Also known as cartilage-hair dysplasia, the McKusick type is most commonly discovered among the many Amish population in Lancaster, Pennsylvania, however it may also be found outside this neighborhood. They may need a change in T-cell immunity, which causes an elevated threat of infection, especially varicella zoster (217, 218). Anemia occurs incessantly in young youngsters, however the incidence lessens as they turn into adolescents (219). A high threat of malignancies, similar to lymphoma, sarcoma, and pores and skin cancers, could be seen in this patient population (217, 220), and life expectancy is decreased, warranting medical remedy into adulthood more than with many of the different dysplasias. In addition, individuals with McKusick-type dysplasia have generalized ligamentous laxity of the elbows and may develop substantial instability although they could have flexion contractures. Bracing is ineffective in controlling the genu varum in these patients, so if it is progressive and painful, operative intervention is warranted. Photograph of a 24-year-old Amish female with McKusick-type dysplasia and gentle genu varum. Early statement of scoliosis is warranted; if the scoliosis is progressive or reaches more than forty five levels, posterior spinal fusion is indicated. Traditional therapy can be used with good success, but it must be applied early and aggressively (217). Lower extremity malalignment can be corrected whether it is progressive or causing pain. Later on in life, if premature arthritis develops, complete hip and complete knee arthroplasty can be carried out safely with good outcomes. These genes produce transmembrane glycoproteins that have an effect on cell signaling, interact with fibroblast progress factor, and affect endochondral development and maturation (226). The features turn into progressively more pronounced till maturity, at which level exostoses should cease to develop. The metaphyses are circumferentially enlarged throughout the body, not only in the areas by which there are obvious exostoses. This enlargement offers the kid a quite "stocky" appearance, which is then further exaggerated by the looks of the exostoses. The exostoses may trigger soreness after they come up beneath tendons or in an space vulnerable to contusion, such because the knee or shoulder.
Order generic revectina from indiaRupture of the gravid proglottid releases 40000 eggs, that are excreted in canine feces. Ingestion of eggs typically happens after the dealing with of an infected canine or the ingesting of contaminated water. The ingested embryo, after release from the egg in the duodenum, penetrates the intestinal mucosa and enters the portal circulation. Although in grownup series involvement of the liver occurs three times more incessantly than the lung, involvement of the lung is famous frequently in youngsters. Other sites of infection in approximately 10% of children embrace the brain, bones, genitourinary tract, eyes, spleen, and heart. Hepatic involvement is marked by the development of "cysts" inside the hepatic parenchyma, most frequently inside the proper lobe. Hydatid sand, composed of separated brood cysts and protoscolices, floats inside the principle cyst cavity. Symptoms of echinococcal cyst formation happen as a outcome of cyst progress and subsequent compression of surrounding tissues. Therefore, right higher quadrant pain and fullness could be the solely presenting options. Jaundice could occur as a end result of compression of the porta hepatis; cholangitis may arise secondary to cyst rupture into the biliary tract. Laboratory knowledge are typically non-specific; elevation of serum alkaline phosphatase and aminotransferases could additionally be famous. Radiography of the abdomen may show calcification of the cyst wall in adults; this modification is seldom obvious in children. Ultrasound is helpful and may show the presence of hydatid sand as well as delineate septations and the presence of daughter cysts. Endoscopic retrograde cholangiography might demonstrate involvement of the biliary tree by daughter cysts following rupture of the primary hepatic cyst. Large lesions require should still require cyst decompression, irrigation with scolicidal solutions, and, in some cases, omentoplasty [64]. Laparoscopic approaches have additionally been utilized efficiently; care is taken to keep away from peritoneal dissemination at surgery. Daughter cysts within the biliary tree could additionally be removed endoscopically after sphincterotomy [65]. In unapproachable hepatic lesions, therapy with albendazole, 15 mg/kg daily for 1 to 6 months, has in some circumstances resulted in reduction in cyst dimension. Ascariasis Human an infection with the roundworm Ascaris lumbricoides is extremely common in tropical and temperate regions worldwide. Infection happens by way of ingestion of embryonated eggs handed in human feces and deposited in soil, the place growth into the infective form occurs. As a outcome, incidence is highest among children and in areas with poor sanitation services. Larva penetrating the small bowel mucosa, are carried within the venous circulation to the lungs, and subsequently pass via the lungs and into the esophagus, as quickly as once more reaching the small bowel the place maturation takes place. Mild hepatic abnormalities may be related to migrating larvae; useless larvae might stimulate granuloma formation. Most clinically related hepatic involvement, however, happens when a number of grownup worms pass via the ampulla of Vater into the frequent bile duct. Symptoms of biliary ascariasis, notably extra common in kids than in adults, embrace (in uncomplicated cases) the acute onset of proper higher quadrant pain (100%), vomiting (96%), history of worm infestation (64%), worm passage in stool or vomitus (50%), and fever (27%). Signs include proper upper quadrant tenderness (100%), palpable gallbladder (11%), hepatomegaly (16%), and jaundice (2%) [68]. Complications of biliary ascariasis are rare however might occur in a higher percentage of affected adults (53%) than kids (5%) [68]. Jaundice, hepatomegaly, and fever occur in higher proportions of sophisticated infestations. Death of ascarids inside the frequent bile duct causes mucosal destruction and fibrosis, resulting in stricture and predisposing to stone formation. Abscess formation may happen; rupture of abscesses into the peritoneal or pleural cavities may then observe. Other reported complications embody cholecystitis, perforation of the widespread bile duct, and pylephlebitis of the hepatic and/or portal veins. Laboratory information in uncomplicated biliary ascariasis are regularly unrevealing, with normal serum aminotransferases famous in 90% of patients [68]. Hyperamylasemia may be current in approximately 25% of patients with frequent duct ascarids. Ultrasound could show the presence of ascarids within the frequent duct; abscess formation could also be noted. Definitive prognosis may be made by demonstration of grownup worms or ova in stool or vomitus. Panendoscopy might reveal the presence of ascarids within the duodenum, while endoscopic retrograde cholangiography permits demonstration of adult worms throughout the biliary tree. Therapy of biliary ascariasis in uncomplicated inections is expectant; antihelminthic remedy consists of albendazole, mebendazole, or ivernectin. In resistant infections, endoscopic sphincterotomy with worm extraction may be effective [69]. Nasobiliary tube placement may enable instillation of antihelminthic agents into the biliary system. Surgery could additionally be required when hepatic abscess is current, in addition to in the presence of common duct and gallbladder perforation. Toxocariasis Visceral larva migrans, brought on by the nematode Toxocara canis, happens worldwide. After ingestion by a dog, embryonated ova endure a life cycle just like that described above for A. Additionally, fetal animals could also be infected in utero through transplacental unfold of the organism. Human an infection typically outcomes from ingestion of soil contaminated by embryonated ova. Subsequent to ingestion, larvae penetrate the intestinal wall and migrate through lymphatics and the venous circulation to , mostly, the liver and lung. Hepatosplenomegaly, rales/wheezing, lymphadenopathy, and pruritic skin lesions could additionally be present on bodily examination. Marked eosinophilia and hyperglobulinemia are generally present, while routine biochemical indicators of hepatic injury are typically normal. Early pathologic findings in the liver encompass larvae surrounded by eosinophils; later findings embody granulomas composed of epithelioid cells, big cells, lymphocytes, and fibroblasts [70]. Treatment entails the administration of albendazole or mebendazole but the efficacy of these regimens stays uncertain. Corticosteroids could also be utilized concomitantly in kids with pulmonary and/ or ocular illness. Capillariasis Infection of the human liver by Capillaria hepatica, a nematode often affecting rodents, cats, and canine, is uncommon however could additionally be associated with a spectrum of clinical findings much like that famous for toxocariasis.
Purchase revectina cheap onlineThis schema has been further expanded into a genotypic classification of mitochondrial hepatopathies (Table 35. Mutations in nuclear genes coding for non-respiratory chain mitochondrial proteins can also trigger mitochondrial cytopathies. The identification of pathologic mutations in many extra nuclear genes regulating mitochondrial operate are the primary focus of exhaustive research efforts within the area of mitochondrial drugs that will actually lead to new genetic diseases among the over 1000 nuclear-encoded mitochondrial proteins [1]. These often present as neonatal liver failure (either a single life-threatening event or recurrent episodes); as a gradually progressive liver disease that may suddenly deteriorate in early childhood, incessantly related to neuromuscular signs; or as a persistent fibrosing liver disease resulting in portal hypertension and its issues. In the secondary disorders, hepatic mitochondria bear damage or perform is impaired secondary to one other pathologic course of. Among these problems are illnesses of uncertain etiology however clearly involving hepatic mitochondria. Reye syndrome); situations attributable to mitochondrial toxins, medication or metals; and different situations during which mitochondrial lipid peroxidation, microauthophagy, and/or abnormal electron transport have been observed and may be concerned within the pathogenesis of liver dysfunction. The reader is referred to other chapters for detailed discussions in regards to the deficiencies of other particular mitochondrial enzyme techniques. Primary mitochondrial hepatopathies the liver and the gastrointestinal tract are among the major goal organs in inherited defects of mitochondrial perform. Neonatal liver failure One of the extra frequent shows of respiratory chain defects in childhood is that dominated by severe liver failure within the first weeks to months of life. Symptoms include lethargy and hypotonia, vomiting and a poor suck from start, seizures (sometimes subtle), and failure to thrive. In others, in an apparently normal toddler, a viral infection or some other undefined inciting event triggers hepatic and, generally, neurologic deterioration. The key biochemical options in most of those infants are the markedly elevated plasma lactate focus, an elevated molar ratio of plasma lactate to pyruvate (>25 and frequently >30 mol/mol), and elevation of -hydroxybutyrate and the arterial ketone body ratio of -hydroxybutyrate to acetoacetate (>2. The lactic acidosis may worsen during the provision of intravenous glucose, a paradoxical discovering that ought to enhance suspicion of a respiratory chain defect. Antenatal manifestations are frequent in infants affected by respiratory chain problems. These findings suggest that metabolism is perturbed long before the toddler is born. Liver histology shows predominantly microvesicular (sometimes mixed with macrovesicular) steatosis, canalicular cholestasis with bile duct thrombi and bile ductular proliferation, and, in some instances, hepatocellular cholestasis. Periportal and centrilobular fibrosis could also be in depth, with loss of broad bands of hepatocytes inflicting a micronodular cirrhotic appearance. Ultrastructural proof of mitochondrial injury could additionally be observed as swollen mitochondria, abnormal cristae, paracrystalline arrays, and a fluffy matrix, although normal mitochondrial morphology may be present. Mitochondria density may be increased in each hepatocyte, as commonly observed in muscle of sufferers with mitochondrial myopathies, presumably as a compensatory mechanism for impaired perform of particular person mitochondria. These infants may progress rapidly from onset of symptoms to death from liver failure, aspiration or sepsis in the first months of life despite supportive remedy [24]. Unfortunately, most sufferers have extreme neurologic involvement in infancy with a weak cry, poor suck, hypotonia, recurrent apnea, myoclonic epilepsy, or a mixture of these conditions, which should preclude consideration for liver transplantation [19]. Others have developed progressive neuromuscular signs following liver transplant. We have efficiently given a transplant to one such infant with cytochrome c oxidase deficiency who has proven no obvious neuromuscular, cardiac, or ocular involvement during 14 years of follow-up. Other more variable parts of the neonatal presentation embody intrauterine development retardation, hydrops fetalis, neonatal ascites, hypoalbuminemia, elevated -fetoprotein, and renal tubular dysfunction [24]. Neonatal liver failure with multivisceral involvement has also been linked to a de novo homoplasmic mutation in the mitochondrial gene for cytochrome b. The gene product is believed to transfer copper from a chaperone to a subunit of cytochrome c oxidase. In different circumstances, the use of valproic acid to deal with myoclonic seizures has seemingly precipitated hepatic failure, even when no prior liver involvement was evident. Liver histology showed microvesicular steatosis, periportal fibrosis with cholangiolar proliferation, extreme cholestasis, hemosiderosis, and pseudoacinar transformation of hepatocytes. If infants survive long sufficient, typical features of AlpersΈuttenlocher disease may become evident (see below). This gene encodes for a mitochondrial translation elongation issue S and might present with intrauterine development retardation, severe lactic acidosis, hypotonia, renal tubulopathy, cholestasis, and liver failure within the first months of life. Initial stories stressed the myopathic presentation in infancy or later in childhood. Symptoms include vomiting and extreme gastroesophageal reflux, failure to thrive, and developmental delay [19]. Biopsy at presentation with diarrhea, weak point, failure to thrive, and elevated aspartate aminotransferase, alanine aminotransferase, worldwide normalized ratio, and plasma lactate. Biopsy reveals delicate hepatocyte swelling with scattered microvesicular steatosis and cholestasis. In most reported patients, neurologic abnormalities developed prior to dying, although the preliminary hypotonia might have been attributed to the lactic acidosis [25]. Death often happens from liver failure, sepsis, bleeding, or aspiration by 1 yr of age. Eventually lack of hepatocyte mass, cholangiolar proliferation (bile ductular reaction), and portal fibrosis develop. Virtually all mitochondria are abnormal with enlargement and pleomorphic measurement and shape, uncommon cristae, and flocculent matrix. This gene encodes an inner mitochondrial membrane protein of still unsure operate, though it could be involved within the metabolism of reactive oxygen species. Mitochondrial-toxic anticonvulsants may set off or worsen a mitochondrial dysfunction and may precipitate to hepatic failure. The onset of signs generally happens between three months and eight years of life with a second peak between 17 and 24 years of life and is characterized by hepatomegaly and jaundice, with hepatic failure evolving over time. In most of these youngsters, liver failure is preceded by the event of hypotonia, feeding difficulties, signs of gastroesophageal reflux or intractable vomiting, failure to thrive, and ataxia adopted by the onset of comparatively refractory partial motor epilepsy or multifocal myoclonus. The seizure dysfunction may necessitate the usage of multiple anticonvulsants, together with valproic acid, which may trigger liver failure in these sufferers inside months by way of its effects on mitochondrial respiratory chain enzyme activity. In other children, the neurologic features are less severe or with considerably later onset. This medical presentation has additionally been called the AlpersΈuttenlocher syndrome (Alpers progressive infantile poliodystrophy). Striking microscopic modifications within the brain include spongiosis, neuronal loss, and astrocytosis, which progress down through the cortical layers and contain the basal ganglia, cerebellum, and brainstem [31]. However, hepatic decompensation may ensue rapidly and lead to dying in 1Ͳ months. In those that present marked deterioration following the beginning of remedy for seizures, demise has been attributed to hepatotoxicity brought on by the valproate; nonetheless, therapy with this drug may have only accelerated the natural historical past of the disease.
Cheap revectina 3 mg amexThe operate of these proteins is to store copper for subsequent metabolic needs of the hepatocyte, to bind and detoxify extra copper, and to provide copper to chaperones that help in incorporating it into important proteins which are secreted. Important hepatic copper metalloenzymes embody superoxide dismutase (32 kDa), mitochondrial monoamine oxidase (195 kDa), cytochrome c oxidase (290 kDa), and ceruloplasmin. This process represents the main homeostatic mechanism for copper metabolism in people. Copper conjugated to glutathione is a minor pathway of copper excretion into bile. Metallochaperones play a vital key role in copper homeostasis in mammalian (and yeast) cells. These proteins management delivery of copper to specific intracellular targets, by which copper is included into synthesis of important enzymes and proteins [7]. Ceruloplasmin, a blue-colored copper-containing 2globulin (134 kDa), is synthesized mainly by hepatocytes and secreted into the systemic circulation [10]. Its major position as a ferroxidase is to promote iron mobilization from tissues by oxidizing ferrous iron for transfer into transferrin. Ceruloplasmin may also transport copper to different tissues and act as an oxidase towards aromatic amines. The regular plasma concentration of ceruloplasmin in older children and adults, measured by the oxidase enzymatic assay, is 20ʹ5 mg/dL and copper is 70ͱ50 g/dL. This is in distinction to the parturient mother, whose levels are 409 mg/dL and 118ͳ02 g/dL, respectively [13]. It must be noted that the classic assays for ceruloplasmin had been based mostly on its oxidase activity in vitro and, hence, relied on the presence of copper within the molecule [14]. As a outcome, many patients with Wilson disease may now have low normal plasma concentrations of ceruloplasmin measured by the immunoassays. This might have an effect on the predictive worth of ceruloplasmin levels in diagnosing Wilson disease. Copper sure to albumin and amino acids, not together with that contained in ceruloplasmin, is known as "non-ceruloplasminbound copper" or "free copper," which normally totals about 10% of whole plasma copper, with the remainder being accounted for by non-exchangeable copper certain to ceruloplasmin [15]. When copper accumulates in the liver in Wilson illness, or if severe liver damage occurs, copper is released into the circulation and increases the fraction of "free copper" relative to the copper certain to ceruloplasmin. Exocystosis of lysosomal copper throughout the canalicular membrane is the most important source of biliary copper, which is then complexed to massive proteins. Urine copper (<100 g daily) is neither indicative of dietary intake nor essential in copper homeostasis underneath normal circumstances. Pathologic processes that intrude with biliary excretion of copper, corresponding to intrahepatic and extrahepatic hepatobiliary cholestatic disorders, produce copper retention in the liver, usually within the lysosomal fraction of hepatocytes, and are associated with elevated plasma copper and ceruloplasmin levels. In reality, hepatic concentrations of copper in intrahepatic cholestatic issues of childhood and primary biliary cirrhosis in adults could equal or exceed levels present in Wilson disease. Whether or not excess hepatic copper in cholestasis is hepatotoxic stays under debate. The organ distribution of the 80ͱ00 mg of copper in an adult includes 15% within the liver, with lesser quantities in mind, heart, and kidneys in decreasing order. In fetal liver, copper concentration is several-fold larger than that current in older children and adults, most of which is sure to metallothionein in hepatocyte lysosomes [20]. During fetal life, the quantity of copper present within the liver declines from over 90% of total body copper throughout early improvement to roughly 50Ͷ0% at birth. After delivery, hepatic copper content material falls rapidly, reaching grownup ranges by three months of age. Increased blood ranges of thyroxine, estrogens, and testosterone [20] are associated with increased plasma ceruloplasmin and copper, making laboratory values tough to interpret. Insufficiency of corticosteroids decreases biliary copper excretion leading to elevated plasma copper and ceruloplasmin ranges. Mechanisms of copper toxicity High levels of orally or parenterally administered copper accumulate within the liver in most mammalian species. Excess hepatic copper has been shown to cause liver injury in rodents, chickens, ruminants, sheep, Bedlington terriers, and humans [15]. The new child human has the capability to tolerate 5- to 100-fold the hepatic copper content of regular adults [21]. Hepatic lysosomal sequestration of extra copper can also be an efficient mechanism to render non-toxic huge concentrations of intracellular copper. However, in some species, natural accumulation of dietary copper results in extreme liver harm and demise, as observed in sheep and the Bedlington terrier [22]. Many cytosolic enzymes that contain sulfhydryl groups could also be inhibited in vitro by copper [23]. Copper inhibits polymerization of tubulin, the chief protein of microtubules, presumably perturbing intracellular trafficking of proteins and mitotic spindle formation. Copper also capabilities as a pro-oxidant, catalyzing the transformation of hydrogen peroxide to the hydroxyl free radical, which, in turn, may react with and injury polyunsaturated fatty acid residues of cell membranes, thiol-rich proteins, and nucleic acids [23]; it may additionally activate intrinsic apoptotic cell dying pathways. These effects may result in disturbances in plasma membrane perform, mitochondrial oxidative phosphorylation, nuclear control of cell processes, protein synthesis by endoplasmic reticulum, and leakage of lysosomal enzymes into the cytosol. There is considerable proof that oxidative injury is a key think about copper toxicity. Lipid peroxidation has been documented in both hepatic lysosomes and mitochondria isolated from copper-overloaded rats [26]. Moreover, hepatic mitochondria isolated from copper-overloaded rats have abnormal respiration and diminished mitochondrial activity of cytochrome c oxidase in conjunction with increased lipid peroxidation [26]. Metallothionein has been shown to operate as an antioxidant; subsequently, it might not only chelate extra copper however may also play a job in reducing oxidative stress stimulated by copper. Other research are consistent with hepatocyte apoptosis being the primary mode of cell dying in copper toxicity [28], presumably explaining the characteristic gentle elevation of serum aminotransferases related to Wilson disease. Epidemiology It has long been recognized that Wilson disease was transmitted by autosomal recessive inheritance, and so consanguinity is comparatively widespread in affected families. Wilson illness is ubiquitous with a worldwide prevalence of approximately 1 in 30 000, and the heterozygote service state has a prevalence of roughly 1 in 90 [1]. There are isolated communities in Japan, Sardinia, and Israel with a higher prevalence. Patients present with a variety of clinical manifestations, including hepatic presentations frequent in childhood and a later-onset predominately neurologic type. Although there are sometimes similarities within the age of onset and scientific findings of Wilson illness in affected siblings, there may be marked differences in organ system involvement and biochemical findings, suggesting that polygenic or environmental components may play a job in expression of the illness. The disease has been described as late as the eighth decade of life and barely earlier than 3 years of age [8]. Presentation in 40Ͷ0% of patients is with primary options in the second decade of life [31]. The the rest of patients comes to medical consideration during the third and fourth decades with a primarily neurologic (34%) or psychiatric (10%) presentation [31] (Table 28. All sufferers have liver involvement, although it might be asymptomatic and nicely compensated. Although signs attributable to Wilson disease might begin in childhood, the analysis is most likely not made for several years or even decades because of a low index of Wilson disease (hepatolenticular degeneration) History In 1912, Kinnear Wilson, an American neurologist, described the degenerative disease of the central nervous system associated with cirrhosis that now bears his name [29].
References - Wu EQ, Birnbaum H, Marynchenko M, et al: Employees with overactive bladder: work loss burden, J Occup Environ Med 47:439n446, 2005.
- Dalakas MC, Illa I, Dambrosia JM. A controlled trial of high- dose intravenous immune globulin infusions as treatment for dermatomyositis. NEJM 1993; 329: 1993n2000.
- NiGuessan G, Stephens FD: Congenital superior ectopic (thoracic) kidney, Urology 24:219n228, 1984.
|
|


