"Buy azitrin overnight, treatment for uti resistant to cipro."By: J. Matthew Brennan, MD - Associate Professor of Medicine
- Member in the Duke Clinical Research Institute
 https://medicine.duke.edu/faculty/j-matthew-brennan-md
Azitrin 100mg with visaBecause of the mild natriuresis induced by Na+ channel inhibitors, these medication seldom are used as sole brokers in remedy of edema or hypertension; their major utility is together with different diuretics. Coadministration of a Na+ channel inhibitor augments the diuretic and antihypertensive response to thiazide and loop diuretics. More necessary, the power of Na+ channel inhibitors to cut back K+ excretion tends to offset the kaliuretic effects of thiazide and loop diuretics and to end in normal plasma K+ values. Liddle syndrome (described later in this chapter) can be handled effectively with Na+ channel inhibitors. Aerosolized amiloride has been proven to improve mucociliary clearance in patients with cystic fibrosis. By inhibiting Na+ absorption from the surfaces of airway epithelial cells, amiloride augments hydration of respiratory secretions and thereby improves mucociliary clearance. Therapeutic Uses essentially the most harmful antagonistic impact of renal Na+ channel inhibitors is hyperkalemia, which could be life threatening. Consequently, amiloride and triamterene are contraindicated in sufferers with hyperkalemia, in addition to in sufferers at elevated danger of developing hyperkalemia. Routine monitoring of the serum K+ level is important in sufferers receiving K+-sparing diuretics. Cirrhotic sufferers are prone to megaloblastosis due to folic acid deficiency, and triamterene, a weak folic acid antagonist, may increase the likelihood of this adverse event. Triamterene also can scale back glucose tolerance and induce photosensitization and has been associated with interstitial nephritis and renal stones. The commonest opposed effects of amiloride are nausea, vomiting, diarrhea, and headache; these of triamterene are nausea, vomiting, leg cramps, and dizziness. Such combos end in increased mobilization of edema fluid whereas causing lesser perturbations of K+ homeostasis. Owing to its 9,11-epoxide group, eplerenone has very low affinity for progesterone and androgen receptors (<1% and <0. High spironolactone concentrations can intrude with steroid biosynthesis by inhibiting steroid hydroxylases, but these results have restricted medical relevance. Spironolactone, but not eplerenone, is extensively thought of to be an antiandrogenic compound and has been used to deal with hirsutism and acne; however, proof for efficacy is weak (Brown et al. Biochemical research advised that spironolactone is a partial agonist of androgen receptors (Nird� et al. Indeed, a recent case report described spironolactone-induced worsening of prostate cancer attributed to androgen receptor stimulation (Sundar and Dickinson, 2012). Urinary Na+ excretion increases with nesiritide, but the effect could also be attenuated by upregulation of Na+ reabsorption in upstream segments of the nephron. Therefore, these medicine are contraindicated in patients with hyperkalemia and in these at increased danger of growing hyperkalemia. Salicylates may cut back the tubular secretion of canrenone and decrease diuretic efficacy of spironolactone. Owing to its affinity for other steroid receptors, spironolactone could cause gynecomastia, impotence, decreased libido, and menstrual irregularities. Spironolactone additionally may induce diarrhea, gastritis, gastric bleeding, and peptic ulcers (the drug is contraindicated in patients with peptic ulcers). Toxicity, Adverse Effects, Contraindications, Drug Interactions Nesiritide could cause hypotension, and there are considerations about opposed renal effects. Pamabrom is the diuretic ingredient in several over-the-counter merchandise marketed for relief of premenstrual syndrome. Little is understood concerning the pharmacology, diuretic mechanism of action, and efficacy of pamabrom. Adenosine Receptor Antagonists There are 4 adenosine receptor subtypes (A1, A2A, A2B, and A3). The A1 receptor is expressed in the proximal tubule and stimulates reabsorption of Na+. Consequently, antagonists of A1 receptors trigger diuresis/natriuresis, yet are K+ sparing. Over any given time interval, the net change in total-body Na+ is the dietary Na+ intake minus the urinary excretion fee and other losses (lower left). Administering diuretics to left-shift the renal pressurenatriuresis relationship; C. CrCl indicates creatinine clearance in milliliters per minute, and ceiling dose refers to the smallest dose of diuretic that produces a near-maximal effect. The scientific situation dictates whether or not a patient ought to receive diuretics and what therapeutic regimen should be used (type of diuretic, dose, route of administration, and velocity of mobilization of edema fluid). Massive pulmonary edema in sufferers with acute left-sided heart failure is a medical emergency requiring speedy, aggressive remedy, including intravenous administration of a loop diuretic. Conversely, gentle pulmonary and venous congestion related to chronic heart failure is best handled with an oral loop or thiazide diuretic, the dosage of which ought to be titrated fastidiously to maximize the benefit-to-risk ratio. Loop and thiazide diuretics lower morbidity and mortality in sufferers with heart failure (Faris et al. Periodic administration of diuretics to cirrhotic patients with ascites might eliminate the need for or cut back the interval between paracenteses, including to affected person consolation and sparing protein reserves which are misplaced during paracenteses. Although diuretics can scale back edema related to continual renal failure, elevated doses of more highly effective loop diuretics often are required. In such cases, solely partial removal of edema fluid ought to be attempted, and fluid must be mobilized slowly utilizing a diuretic regimen that accomplishes the duty with minimal perturbation of regular physiology. If diuretic resistance develops towards a less-efficacious diuretic, a extra efficacious diuretic should be substituted, such as a loop diuretic for a thiazide. Consequently, diuretic focus on the active website in the tubular lumen is diminished. In nephrotic syndrome, binding of diuretics to luminal albumin was postulated to restrict response; nevertheless, the validity of this concept has been challenged. In hepatic cirrhosis, nephrotic syndrome, and heart failure, nephrons might have diminished diuretic responsiveness due to increased proximal tubular Na+ reabsorption, resulting in diminished Na+ delivery to distal nephrons. Faced with resistance to loop diuretics, the clinician has a number of options: � Bed rest could restore drug responsiveness by improving the renal circulation. For instance, a combination of a loop diuretic with a K+-sparing or a thiazide diuretic might improve therapeutic response; nevertheless, nothing is gained by the administration of two drugs of the same type. The hormone is launched by the posterior pituitary whenever water deprivation causes an elevated plasma osmolality or whenever the cardiovascular system is challenged by hypovolemia or hypotension. Vasopressin acts primarily in the renal amassing duct to increase the permeability of the cell membrane to water, thus permitting water to move passively down an osmotic gradient throughout the amassing duct into the extracellular compartment. Vasopressin also promotes release of coagulation factors by vascular endothelium and increases platelet aggregability. Vasopressin synthesis seems to be regulated solely on the transcriptional stage. The prohormone accommodates three domains: vasopressin (residues 1�9), vasopressin-neurophysin (residues 13�105), and vasopressin-glycopeptide (residues 107�145). The synthesis and transport of vasopressin depend on the preprohormone conformation.
Order discount azitrin on linePrecipitants embrace stress, fatigue, emergence into a dark environment, marijuana, antipsychotic brokers, and anxiousness states. Acute effects are dose dependent and embrace feelings of energy, altered sense of time, and nice sensory experiences with enhanced perception. At higher doses, visible hallucinations, agitation, hyperthermia, and panic attacks have been reported. A typical oral dose is one or two 100-mg tablets, producing results lasting 3�6 h, though dosage and potency of street samples are variable (100 mg of lively drug per tablet). It was classified as a dissociative anesthetic as a end result of, within the anesthetized state, the affected person remains conscious with staring gaze, flat facies, and rigid muscles. Abusers taking larger doses could appear to be reacting to hallucinations and should exhibit hostile or assaultive habits. Anesthetic effects enhance with dosage; stupor or coma may happen with muscular rigidity, rhabdomyolysis, and hyperthermia. Intoxicated sufferers within the emergency room might progress from aggressive habits to coma, with elevated blood stress and enlarged, nonreactive pupils. Nicotine absorption and cardiovascular results with smokeless tobacco use: comparability with cigarettes and nicotine gum. Vaccine for cocaine dependence: a randomized doubleblind placebo-controlled efficacy trial. Extended-release naltrexone to forestall opioid relapse in legal justice offenders. Drug dependence, a chronic medical sickness: Implications for remedy, insurance coverage, and outcomes analysis. Modafinil and sleep architecture in an inpatatientoutpatient therapy research of cocaine dependence. Accelerating cocaine metabolism as an strategy to the remedy of cocaine abuse and toxicity. The seek for genes related to a low-level response to alcohol determined by alcohol challenges. A double-blind examine evaluating the long-term security of varenicline for smoking cessation. Drugs Affecting Renal Excretory Function / 445 Renin and Angiotensin / 471 Treatment of Ischemic Heart Disease / 489 Treatment of Hypertension / 507 Therapy of Heart Failure / 527 Antiarrhythmic Drugs / 547 Treatment of Pulmonary Arterial Hypertension / 573 Blood Coagulation and Anticoagulant, Fibrinolytic, and Antiplatelet Drugs / 585 Chapter 33. Disease states such as hypertension, coronary heart failure, renal failure, nephrotic syndrome, and cirrhosis could disrupt this stability. Diuretics improve the rate of urine flow and Na+ excretion and are used to regulate the quantity or composition of physique fluids in these issues. It is managed by a finely tuned homeostatic mechanism that operates by adjusting each the speed of water intake and the rate of solute-free water excretion by the kidneys-that is, water stability. Abnormalities in this homeostatic system can result from genetic ailments, acquired illnesses, or drugs and may trigger critical and doubtlessly life-threatening deviations in plasma osmolality. Part I of this chapter first describes renal physiology, then introduces diuretics with regard to mechanism and site of motion, results on urinary composition, and effects on renal hemodynamics, and then integrates diuretic pharmacology with a dialogue of mechanisms of edema formation and the function of diuretics in medical medicine. Specific therapeutic purposes of diuretics are introduced in Chapters 28 (hypertension) and 29 (heart failure). Glomerular Filtration In the glomerular capillaries, a portion of plasma water is pressured through a filter that has three fundamental parts: the fenestrated capillary endothelial cells, a basement membrane lying simply beneath the endothelial cells, and the filtration slit diaphragms fashioned by epithelial cells that cover the basement membrane on its urinary house aspect. The kidney filters giant portions of plasma, reabsorbs substances that the body must preserve, and leaves behind or secretes substances that should be eliminated. The two kidneys in people together produce about 120 mL of ultrafiltrate/min, but just one mL of urine/min of urine; greater than 99% of the glomerular ultrafiltrate is reabsorbed at a staggering power cost. Normally, about 65% of filtered Na+ is reabsorbed in the proximal tubule, and since this part of the tubule is highly permeable to water, reabsorption is actually isotonic. The macula densa is strategically situated to sense concentrations of NaCl leaving the loop of Henle. If the concentration of NaCl is merely too Overview of Nephron Function Part I: Renal Physiology and Diuretic Drug Action Renal Anatomy and Physiology the essential urine-forming unit of the kidney is the nephron. The glomerulus receives blood from an afferent arteriole, and blood exits the glomerulus through an efferent arteriole. The macula densa additionally regulates renin launch from the adjacent juxtaglomerular cells within the wall of the afferent arteriole. It is right here that final adjustments in electrolyte composition are made, a process modulated by the adrenal steroid aldosterone. The extra distal parts of the accumulating duct move via the renal medulla, where the interstitial fluid is markedly hypertonic. The movement of water out of the tubule is pushed by the steep concentration gradient that exists between tubular fluid and medullary interstitium. Because this phase of the nephron is impermeable to water, energetic transport within the ascending limb dilutes the tubular fluid. Because the cortical and outer medullary accumulating ducts have low permeability to urea, urea is concentrated in the tubular fluid. Na+ can diffuse down this Na+ gradient throughout the luminal membrane by way of Na+ channels and via membrane symporters that use the energy saved within the Na+ gradient to transport solutes out of the tubular lumen and into the cell. Na+ exits the basolateral membrane into intercellular and interstitial spaces through the Na+ pump. The motion of Na+-linked symporters in the luminal membrane causes the focus of substrates for these symporters to rise in the epithelial cell. These substrate/solute gradients then allow simple diffusion or mediated transport. Accumulation of Na+ and different solutes within the intercellular house creates a small osmotic strain differential throughout the epithelial cell. In water-permeable epithelium, water moves into the intercellular areas pushed by the osmotic stress differential. Bulk water circulate carries some solutes into the intercellular space by solvent drag. Movement of water into the intercellular space concentrates other solutes in the tubular fluid, leading to an electrochemical gradient for these substances throughout the epithelium. Membrane-permeable solutes then move down their electrochemical gradients into the intercellular area by each the transcellular. Membrane-impermeable solutes stay in the tubular lumen and are excreted in the urine with an compulsory amount of water. As water and solutes accumulate within the intercellular house, hydrostatic stress increases, thus providing a driving drive for bulk water circulate. Bulk water flow carries solute out of the intercellular house into the interstitial space and, lastly, into the peritubular capillaries. Both techniques are powered by the sodium pump within the basolateral membrane, involve secondary and tertiary active transport, and use a facilitated diffusion step. Renal Handling of Specific Anions and Cations Organic Acid and Organic Base Secretion the kidney is a serious organ involved within the elimination of natural chemicals from the physique. Organic molecules could enter the renal tubules by glomerular filtration or could additionally be actively secreted directly into tubules. The numbers 1, 2, and three refer to major, secondary, and tertiary energetic transport, respectively.
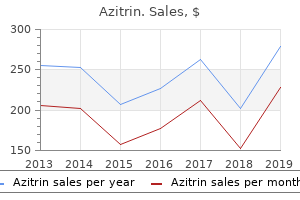
Buy azitrin overnightDuloxetine, along with being permitted to be used within the therapy of melancholy and anxiousness, is used for therapy of fibromyalgia and neuropathic pain related to peripheral neuropathy (Finnerup et al. These embody two close structural analogues, trazodone and nefazodone, as nicely as mirtazapine and mianserin (not marketed in the U. Both mianserin and mirtazapine are quite sedating and are remedies of selection for some depressed sufferers with insomnia. The hydroxybupropion metabolite might contribute to the therapeutic effects of the parent compound: this metabolite seems to have a similar pharmacology and is current at substantial levels. Bupropion is indicated for the remedy of melancholy, prevention of seasonal depressive disorder, and as a smoking cessation therapy (Carroll et al. The mechanism of motion and adverse results of the atypical antipsychotics are described in Chapter sixteen. The main dangers of these brokers are weight achieve and metabolic syndrome, a larger drawback for quetiapine and olanzapine than for aripiprazole. Selegiline is available as a transdermal patch for the remedy of melancholy; transdermal supply could reduce the chance for diet-associated hypertensive reactions. The olanzapine-fluoxetine mixture is available in fixed-dose combos of 3, 6, or 12 mg of olanzapine and 25 or 50 mg of fluoxetine. Likewise, dose concerns need to embrace consciousness of hepatic function (Mauri et al. Both immediate-release and extended-release (tablet or capsule) preparations of venlafaxine result in steady-state levels of drug in plasma within 3 days. The elimination half-lives for the parent venlafaxine and its lively and major metabolite desmethylvenlafaxine are 5 and eleven h, respectively. Venlafaxine dose reductions are instructed for patients with renal or hepatic impairment. The recommended preliminary dosing of mirtazapine is 15 mg/d, with a maximal beneficial dose of 45 mg/d. Clearance of mirtazapine is decreased within the aged and in sufferers with moderate-to-severe renal or hepatic impairment. Pharmacokinetics and opposed results of mirtazapine could have an enantiomer-selective element (Brockm�ller et al. Trazodone typically is started at 150 mg/d in divided doses, with 50-mg increments every 3�4 days. The maximally beneficial dose is four hundred mg/d for outpatients and 600 mg/d for inpatients. Nefazodone has a t1/2 of only 2�4 h; its major metabolite hydroxynefazodone has a t1/2 of 1. Bupropion elimination has a t1/2 of 21 h and involves each hepatic and renal routes. Patients with severe hepatic cirrhosis should obtain a most dose of one hundred fifty mg each other day; consideration for a decreased dose also wants to be made in circumstances of renal impairment. Steady-state concentrations happen within several days to a quantity of weeks of starting treatment, as a operate of the t1/2. Nonetheless, monitoring the plasma exposure has an essential relationship to treatment response: There is a relatively slim therapeutic window. To avoid toxicity in "gradual metabolizers," plasma ranges ought to be monitored and doses adjusted downward. Monoamine Oxidase Inhibitors has not yet been proven to have a practical influence on choice of drug therapy in clinical settings (Dubovsky, 2015). In the case of fluoxetine, the mixed action of the mother or father and the demethylated metabolite norfluoxetine permits for a once-weekly formulation. Aspects of sexual dysfunction could be handled in each men and women with the phosphodiesterase 5 inhibitor sildenafil (Nurnberg, 2001; Nurnberg et al. With continued treatment, some sufferers also report a dullness of mental talents and focus. Thus, dosage changes are primarily based more on analysis of medical response and management of unwanted effects. Sudden withdrawal of antidepressants can precipitate a discontinuation syndrome (Harvey and Slabbert, 2014). This withdrawal syndrome seems most intense for paroxetine and venlafaxine because of their comparatively brief half-lives and, in the case of paroxetine, lack of active metabolites. Conversely, the energetic metabolite of fluoxetine, norfluoxetine, has such a long t1/2 (1�2 weeks) that few sufferers expertise any withdrawal symptoms with discontinuation of fluoxetine. The immediate-release formulation of venlafaxine can induce sustained diastolic hypertension (diastolic blood pressure > ninety mm Hg at consecutive weekly visits) in 10%�15% of sufferers at larger doses; this danger is decreased with the extended-release form. Serotonin Receptor Antagonists Regarding the serotonin receptor antagonists, the main unwanted effects of mirtazapine, seen in more than 10% of the patients, are somnolence, elevated urge for food, and weight gain. Nefazodone was voluntarily withdrawn from the market in several nations after uncommon instances of liver failure had been related to its use. Bupropion Typical unwanted effects related to bupropion include anxiety, mild tachycardia and hypertension, irritability, and tremor. Other unwanted facet effects include headache, nausea, dry mouth, constipation, urge for food suppression, insomnia, and, not often, aggression, impulsivity, and agitation. Seizures are dependent on dose and Cp, with seizures occurring not often within the beneficial dose vary. Bupropion ought to be avoided in patients with seizure disorders in addition to those with bulimia as a end result of an increased risk of seizures (Horne et al. At doses larger than that really helpful for despair (450 mg/d), the chance of seizures will increase considerably. The use of extended-release formulations typically blunts the maximum focus noticed after dosing and minimizes the possibility of reaching drug ranges associated with an increased risk of seizures. Antagonism of 1 adrenergic receptors contributes to orthostatic hypotension and sedation. This is the primary reason that only a limited provide ought to be available to the patient at any given time. Tricyclic Antidepressants Values are experimentally determined potencies (Ki values, nM) for binding to receptors that contribute to frequent unwanted effects of clinically used antidepressant drugs: muscarinic cholinergic receptors. The launched catecholamines stimulate postsynaptic receptors within the periphery, increasing blood strain to dangerous ranges. Symptoms of the serotonin syndrome embody hyperthermia, muscle rigidity, myoclonus, tremors, autonomic instability, confusion, irritability, and agitation; this will progress towards coma and dying. The benzodiazepines are effective anxiolytics as both acute and chronic remedy. There is concern regarding their use due to their potential for dependence and abuse as well as unfavorable effects on cognition and reminiscence. Antihistamines and sedative-hypnotic agents have been tried as anxiolytics however are usually not recommended because of their side-effect profiles and the supply of superior medicine. Failure to observe these required waiting periods may end up in the serotonin syndrome. Clinical Considerations With Anxiolytic Drugs the choice of pharmacological therapy of tension is dictated by the precise anxiety-related disorders and the medical want for acute anxiolytic effects (Millan, 2003).
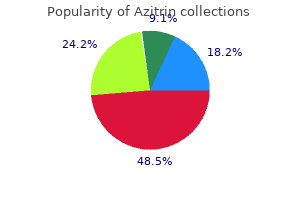
Buy azitrin 100 mg amexIn response to changes in native transmembrane potential, ion channels bear conformational changes, allowing for, or preventing, the circulate of ions via the conducting pore alongside their electrochemical gradient, typically in time-, voltage-, or ligand-dependent trend. To provoke an motion potential, a cardiac myocyte at relaxation is depolarized above a threshold potential, normally by way of hole junctions by a neighboring myocyte. At hyperpolarized potentials, the channel is in a closed conformation, and no present can circulate (left). As depolarization begins, the voltage sensor (indicated here as ++++) moves, thus altering channel conformation and opening the pore, permitting conduction (middle). As depolarization is maintained, an intracellular particle blocks current move, making the channel nonconducting in this inactivated state (right). Na+ current and is a serious determinant of conduction velocity of a propagating action potential. The relationship between Na+ channel availability and transmembrane potential is a vital determinant of conduction and refractoriness in many cells, as discussed in the material that follows. During the part 2 plateau of a normal cardiac action potential, inward, depolarizing currents, primarily through L-type Ca2+ channels, are balanced by outward, repolarizing currents primarily via K+ ("delayed rectifier") channels. The identification of disease genes not solely has resulted in improved care of affected patients and their families but also has contributed importantly to our understanding of the normal action potential, arrhythmia mechanisms, and potential antiarrhythmic drug targets (Keating and Sanguinetti, 2001). Inhibitors could embody not only antiarrhythmics corresponding to mexiletine or flecainide mentioned in this chapter, but in addition the antianginal agent ranolazine (see Chapter 28), which seems to be a late Na+ present blocker. Intriguingly, some arrhythmias in acquired coronary heart illness have been attributed to elevated late Na+ current or leaky RyR2 channels. Thus, research in the rare congenital arrhythmia syndromes could point to new avenues for drug development in more widespread arrhythmias in acquired coronary heart disease (Knollmann and Roden, 2008 Priori et al. The present magnitudes are to not scale; the Na+ present is ordinarily 50 times bigger than any other current, although the portion that persists into the plateau (phase 2) is small. Each represents a different channel protein, usually associated with ancillary (function-modifying) subunits. The genes encoding the main pore-forming proteins have been cloned for most of the channels shown here and are listed within the righthand column. The resultant diversity of motion potentials in different areas of the heart plays a job in understanding the pharmacological profiles of antiarrhythmic medication. In the ventricle, action potential duration varies across the wall of every chamber, in addition to apicobasally, largely as a consequence of varying densities of repolarizing currents. In addition, these cells, in addition to cells from the conducting system, normally show the phenomenon of spontaneous diastolic, or section four, depolarization and thus spontaneously attain threshold for regeneration of motion potentials. The price of spontaneous firing usually is fastest in sinus node cells, which therefore serve as the pure pacemaker of the heart. The gradual diastolic depolarization that underlies pacemaker exercise is generated by a nonselective channel that conducts both Na+ and K+ and is activated at hyperpolarized membrane potentials (Cohen and Robinson, 2006). Certain ion channels are expressed only in some tissues or become energetic solely under particular pathophysiologic conditions. For example, the T-type Ca2+ channel may be essential in ailments corresponding to hypertension and play a task in pacemaker exercise (Ono and Iijima, 2010). A T-type-selective Ca2+ channel antagonist, mibefradil was commercially available briefly within the late Nineteen Nineties but was withdrawn because of concerns over potentially life-threatening pharmacokinetic interactions with many other medication. Impulse Propagation and the Electrocardiogram Normal cardiac impulses originate within the sinus node. Impulse propagation in the heart is determined by the magnitude of the depolarizing current (usually Na+ current) and the geometry and density of cell-cell electrical connections (Kleber and Saffitz, 2014). Cardiac cells are relatively lengthy and thin and properly coupled via specialized gap junction proteins at their ends, whereas lateral ("transverse") gap junctions are sparser. As a outcome, impulses spread alongside cells two to thrice faster than across cells. This "anisotropic" (direction-dependent) conduction could also be an element in the genesis of certain arrhythmias described in the material that follows (Priori et al. Activation spreads from the His-Purkinje system on the endocardium of the ventricles throughout the relaxation of the ventricles, stimulating coordinated ventricular contraction. When a stimulus occurs during section three of the motion potential, the upstroke of the untimely motion potential is slower and of smaller magnitude. Thus, refractoriness is decided by the voltage-dependent recovery of Na+ channels from inactivation. Refractoriness incessantly is measured by assessing whether or not untimely stimuli utilized to tissue preparations (or the whole heart) result in propagated impulses. While the magnitude of the Na+ current is one main determinant of propagation of untimely beats, mobile geometry also is necessary in multicellular preparations. Propagation from cell to cell requires current flow from the primary web site of activation and consequently can fail if inward current is inadequate to drive activation in many neighboring cells. Even after a Ca2+ channel�dependent action potential has repolarized to its initial resting potential, not all Ca2+ channels can be found for reexcitation. Therefore, an additional stimulus utilized shortly after repolarization is full generates a lowered Ca2+ present, which can propagate slowly to adjacent cells previous to extinction. An extra stimulus applied later will end in a larger Ca2+ present and faster propagation. Conduction that exhibits such dependence on the timing of premature stimuli is termed decremental. Slow conduction within the coronary heart, a crucial issue within the genesis of reentrant arrhythmias (see additional discussion), also can occur when Na+ currents are depressed by disease or membrane depolarization. These abnormalities could additionally be attributable to medication (Table 30�1) or by structural heart disease; within the latter case, everlasting cardiac pacing may be required. Abnormally fast heart rhythms (tachyarrhythmias) are widespread clinical issues that might be handled with antiarrhythmic medication. Three major underlying mechanisms have been identified: enhanced automaticity, triggered automaticity, and reentry. These are sometimes interrelated mechanisms as irregular beats arising from one mechanism can elicit a second; for instance, a triggered automated beat can provoke reentry. Adrenergic stimulation, hypokalemia, and mechanical stretch of cardiac muscle cells increase part four slope and so accelerate pacemaker rate, whereas acetylcholine reduces pacemaker rate both by decreasing phase 4 slope and by hyperpolarization (making the utmost diastolic potential more negative). In addition, computerized habits could happen in websites that ordinarily lack spontaneous pacemaker exercise; for example, depolarization of ventricular cells. When impulses propagate from a area of enhanced normal or irregular automaticity to excite the relaxation of the heart, more complicated arrhythmias might outcome from the induction of reentry. If this irregular depolarization reaches threshold, it could, in flip, give rise to secondary upstrokes that may propagate and create irregular rhythms. These abnormal secondary upstrokes happen only after an initial regular, or "triggering," upstroke and thus are termed triggered rhythms. If this afterdepolarization reaches threshold, a secondary triggered beat or beats might occur. In the second sort of triggered exercise, the vital thing abnormality is marked prolongation of the cardiac motion potential. With a very early untimely stimulus (black arrow) in ventricular myocardium, all Na+ channels nonetheless are within the inactivated state, and no upstroke results. As the action potential repolarizes, Na+ channels get well from the inactivated to the resting state, from which opening can happen.
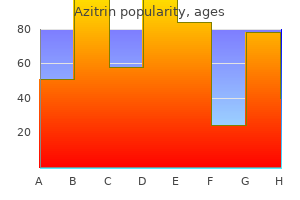
Purchase 500mg azitrin with mastercardSome of the vasoconstrictor brokers may be absorbed systemically, sometimes to an extent adequate to cause untoward reactions (see the following section). There additionally may be delayed wound therapeutic, tissue edema, or necrosis after local anesthesia. These results seem to occur partly as a result of sympathomimetic amines enhance the O2 consumption of the tissue; this, along with the vasoconstriction, results in hypoxia and local tissue damage. Thus, using vasoconstrictors in native anesthetic preparations for anatomical areas with restricted collateral circulation is avoided. Ventricular tachycardia and fibrillation are relatively unusual penalties of local anesthetics apart from bupivacaine. The antiarrhythmic effects of local anesthetics such as lidocaine and procainamide are mentioned in Chapter 30. Finally, it should be stressed that untoward cardiovascular effects of native anesthetic agents may result from their inadvertent intravascular administration, particularly if epinephrine can be present. Smooth Muscle Local anesthetics depress contractions in the intact bowel and in strips of isolated gut (Zipf and Dittmann, 1971). They also chill out vascular and bronchial easy muscle, although low concentrations initially may produce contraction (Covino, 1987). Local anesthetics may improve the resting tone and reduce the contractions of isolated human uterine muscle; nonetheless, uterine contractions are seldom depressed instantly during intrapartum regional anesthesia. The danger of such opposed reactions is proportional to the concentration of native anesthetic achieved in the circulation. In common, in local anesthetics with chiral centers, the S-enantiomer is much less poisonous than the R-enantiomer (McClure, 1996). Neuromuscular Junction and Ganglia Local anesthetics also affect transmission on the neuromuscular junction. In general, the more potent the anesthetic, the more readily convulsions could also be produced. Central stimulation is adopted by depression; demise normally is attributable to respiratory failure. Under these conditions, the focus of the drug probably rises so rapidly that every one neurons are depressed concurrently. Airway control, along with ventilatory and circulatory support, are essential options of treatment in the late stage of intoxication. Intravenously administered benzodiazepines are the drugs of choice for both the prevention and the arrest of convulsions. Neither propofol nor a quickly appearing barbiturate is most well-liked; both usually tend to produce cardiovascular despair than a benzodiazepine (Chapter 19). Whereas different local anesthetics additionally present the effect, cocaine has a particularly prominent impact on temper and behavior. The reaction may manifest itself as an allergic dermatitis or a typical asthmatic attack (Covino, 1987). It is necessary to distinguish allergic reactions from toxic unwanted aspect effects and from the results of coadministered vasoconstrictors. Hypersensitivity appears to happen more regularly with native anesthetics of the ester sort and regularly extends to chemically associated compounds. For instance, individuals delicate to procaine also may react to structurally comparable compounds. Although allergic responses to agents of the amide type are uncommon, options of such brokers could comprise preservatives corresponding to methylparaben that will provoke an allergic reaction (Covino, 1987). Local anesthetic preparations containing a vasoconstrictor also may elicit allergic responses as a outcome of the sulfite added as an antioxidant for the catecholamine/vasoconstrictor. Because spinal fluid accommodates little or no esterase, anesthesia produced by the intrathecal injection of an anesthetic agent will persist until the native anesthetic agent has been absorbed into the circulation. However, with prilocaine, the preliminary step is hydrolytic, forming o-toluidine metabolites that may cause methemoglobinemia. The extensive use of amide-linked native anesthetics in patients with extreme hepatic disease requires caution. Cardiovascular System Following systemic absorption, local anesthetics act on the cardiovascular system. The major web site of action is the myocardium, where decreases in electrical excitability, conduction price, and pressure of contraction occur. However, on rare occasions, decrease doses of some native anesthetics will trigger cardiovascular collapse Toxicity the metabolic fate of local anesthetics is of nice sensible importance as a result of toxicity may finish up from an imbalance between their rates of absorption and elimination. The fee of absorption of many native anesthetics into the systemic circulation may be significantly lowered by the incorporation of a vasoconstrictor agent within the anesthetic solution. However, the speed of degradation of native anesthetics varies tremendously, and this is a vital component in determining the protection of a selected agent. Because toxicity is expounded to the concentration of free drug, binding of the anesthetic to proteins within the serum and to tissues reduces toxicity. For instance, in intravenous regional anesthesia of an extremity, about half of the original anesthetic dose still is tissue sure 30 min after the restoration of normal blood flow (Arthur, 1987). Reversing the results of native anesthetic systemic toxicity is a clinical challenge. One creating method is promising and strange: intravenous lipid emulsion remedy (Weinberg, 2012). The amide-linked native anesthetics bind extensively (55%�95%) to plasma proteins, significantly 1-acid glycoprotein. The neonate is relatively poor in plasma proteins that bind native anesthetics and thereby is extra prone to toxicity. Uptake by the lung also might play an important position in the distribution of amide-linked local anesthetics. Finally, lowered cardiac output slows supply of the amide compounds to the liver, reducing their metabolism and prolonging their plasma half-lives. A lidocaine transdermal patch is used for relief of ache associated with postherpetic neuralgia. Lidocaine together with tetracaine in a formulation that generates a "peel" is permitted for topical local analgesia previous to superficial dermatological procedures similar to filler injections and laser-based remedies. Lidocaine together with tetracaine is also equipped in a formulation that generates warmth on publicity to air, which is used prior to venous entry and superficial dermatological procedures such as excision, electrodessication, and shave biopsy of skin lesions. The gentle warming is meant to enhance pores and skin temperature by as a lot as 5�C for the aim of enhancing delivery of native anesthetic into the pores and skin. In addition to preparations for injection, lidocaine is formulated for topical, ophthalmic, mucosal, and transdermal use. Both monoethylglycine xylidide and glycine xylidide retain local anesthetic exercise. In people, about 75% of the xylidide is excreted in the urine as the additional metabolite 4-hydroxy-2,6-dimethylaniline (Arthur, 1987). Toxicity Local Anesthetics and Related Agents Cocaine Chemistry Cocaine, an ester of benzoic acid and methylecgonine, occurs in abundance in the leaves of the coca shrub. Ecgonine is an amino alcohol base intently associated to tropine, the amino alcohol in atropine.

D-Alpha-Tocopheryl Acetate (Vitamin E). Azitrin. - A type of arthritis called osteoarthritis. Vitamin E does not seem to decrease pain or stiffness and does not seem to prevent osteoarthritis from getting worse.
- Prostate cancer prevention.
- Helping people walk without pain when they have a disease called intermittent claudication.
- Helping the eyes heal after surgery.
- Decreasing sunburn.
- High blood pressure during pregnancy (pre-eclampsia).
- Hot flashes in people who have had breast cancer.
Source: http://www.rxlist.com/script/main/art.asp?articlekey=96917
250 mg azitrin overnight deliveryPrimidone is contraindicated in sufferers with either porphyria or hypersensitivity to phenobarbital. Therapeutic concentrations are reported to be 6�12 g/mL, although appreciable variation occurs. Therapeutic Uses Iminostilbenes Carbamazepine Carbamazepine is taken into account to be a main drug for the remedy of generalized tonic-clonic, focal-to-bilateral tonic-clonic, tonic-clonic of unknown onset (generalized tonic-clonic), and focal seizures. It is a spinoff of iminostilbene with a carbamyl group on the 5 position; this moiety is crucial for potent antiseizure exercise. Carbamazepine is helpful in patients with generalized tonic-clonic and both focal conscious and focal with impaired awareness seizures (Table 17�1). The therapeutic use of carbamazepine is discussed further on the finish of this chapter. Carbamazepine is the first agent for treatment of trigeminal and glossopharyngeal neuralgias. It can be efficient for lightning-type ("tabetic") ache associated with bodily losing. Carbamazepine is also used within the treatment of bipolar affective issues, as discussed additional in Chapter sixteen. During longterm therapy, the extra frequent untoward results of the drug embrace drowsiness, vertigo, ataxia, diplopia, and blurred vision. Other antagonistic effects embody nausea; vomiting; serious hematological toxicity (aplastic anemia, agranulocytosis); and hypersensitivity reactions (dangerous skin reactions, eosinophilia, lymphadenopathy, splenomegaly). A late complication of remedy with carbamazepine is retention of water, with decreased osmolality and concentration of Na+ in plasma, especially in aged sufferers with cardiac disease. Some tolerance develops to the neurotoxic effects of carbamazepine, and they are often minimized by gradual improve in dosage or adjustment of upkeep dosage. Various hepatic or pancreatic abnormalities have been reported throughout remedy with carbamazepine, most commonly a transient elevation of hepatic transaminases in plasma in 5%�10% of sufferers. A transient, gentle leukopenia occurs in about 10% of patients throughout initiation of remedy and usually resolves throughout the first four months of continued remedy; transient thrombocytopenia also has been famous. In about 2% of patients, a persistent leukopenia might develop that requires withdrawal of the drug. The initial concern that aplastic anemia may be a frequent complication of long-term remedy with carbamazepine has not materialized. In most cases, the administration of a quantity of drugs or the presence of another underlying illness has made it difficult to set up a causal relationship. Concurrent administration of carbamazepine may decrease concentrations of valproate, lamotrigine, tiagabine, and topiramate. Carbamazepine reduces both the plasma concentration and the therapeutic impact of haloperidol. The metabolism of carbamazepine may be inhibited by propoxyphene, erythromycin, cimetidine, fluoxetine, and isoniazid. Eslicarbazepine competitively inhibits fast voltage-gated sodium channels, stabilizing the inactivated state and the sodium-dependent launch of neurotransmitters. Succinimides Ethosuximide Ethosuximide is a primary agent for the remedy of generalized absence seizures. The thalamus performs an necessary role in era of 3-Hz spike-and-wave rhythms typical of absence seizures (Huguenard and McCormick, 2007). Neurons within the thalamus exhibit large-amplitude T-type currents that underlie bursts of motion potentials and sure play an important position in thalamic oscillatory activity, such as 3-Hz spikeand-wave activity. Ethosuximide reduces this current with out modifying the voltage dependence of steady-state inactivation or the time course of restoration from inactivation. Absorption of ethosuximide seems to be complete, with peak Cp occurring inside about 3 h after a single oral dose. The main metabolite, the hydroxyethyl by-product, accounts for about 40% of ethosuximide metabolism, is inactive, and is excreted as such and because the glucuronide in the urine. The plasma t1/2 of ethosuximide averages between 40 and 50 h in adults and about 30 h in youngsters. Eslicarbazepine is then extensively converted to its S(+) enantiomer, the energetic metabolite S-licarbazepine. Oxcarbazepine is inactivated by glucuronide conjugation, is eradicated by renal excretion, and has a brief t1/2 of only about 1�2 h. Oxcarbazepine has a mechanism of action just like that of carbamazepine but is a less-potent enzyme inducer than carbamazepine. Substitution of oxcarbazepine for carbamazepine is associated with increased ranges of phenytoin and valproate, presumably because of reduced induction of hepatic enzymes. Although most antagonistic effects are just like that with carbamazepine, hyponatremia may happen more commonly with oxcarbazepine than with carbamazepine. Plasma Drug Concentrations During long-term remedy, the plasma concentration of ethosuximide averages about 2 g/mL per daily dose of 1 mg/kg. A plasma focus of 40�100 g/mL often is required for passable control of absence seizures. Therapeutic Uses Ethosuximide is efficient towards absence seizures, but not tonic-clonic seizures. An initial day by day dose of 250 mg in kids (3�6 years old) and 500 mg in older youngsters, and grownup dosage is increased by 250-mg increments at weekly intervals till seizures are adequately controlled or toxicity intervenes. Divided dosage is required occasionally to stop nausea or drowsiness related to once-daily dosing. Increased caution is required if the day by day dose exceeds 1500 mg in adults or 750�1000 mg in children. The therapeutic use of ethosuximide is mentioned further at the finish of the chapter. Restlessness, agitation, anxiety, aggressiveness, inability to focus, and other behavioral effects have occurred primarily in sufferers with a prior history of psychiatric disturbance. Urticaria and different pores and skin reactions, including Stevens-Johnson syndrome, systemic lupus erythematosus, eosinophilia, leukopenia, thrombocytopenia, pancytopenia, and aplastic anemia, also have been attributed to the drug. The leukopenia could additionally be transient regardless of continuation of the drug, but several deaths have resulted from bone marrow depression. The potential for such critical and life-threatening antagonistic results has restricted the scientific utility of felbamate. Other Antiseizure Drugs Acetazolamide Acetazolamide, the prototype for the carbonic anhydrase inhibitors, is mentioned in Chapter 25. Its antiseizure actions have been discussed in earlier editions of this textbook. This dual action on excitatory and inhibitory transmitter responses might contribute to the extensive spectrum of action of the drug in seizure models; however, the mechanism(s) by which felbamate exerts its anticonvulsant activity remain unknown. Ezogabine Therapeutic Use Ezogabine is a first-in-class K+ channel opener, generally recognized as retigabine within the E. Ezogabine is metabolized by glucuronidation and acetylation and has a t1/2 of 7�11 h; it and its metabolites are excreted within the urine. Concomitant administration of phenytoin or carbamazepine might reduce plasma concentrations of ezogabine; consequently, an increase in ezogabine dosage should be thought-about when including phenytoin or carbamazepine. Despite the potential serious opposed results, felbamate is used at doses ranging from 1 to four g/d.
Order online azitrinIn addition to morphine, codeine, and the semisynthetic derivatives of the pure opium alkaloids, a variety of other structurally distinct chemical courses of medicine have pharmacological actions much like these of morphine. Clinically helpful compounds embody the morphinans, benzomorphans, methadones, phenylpiperidines, and propionanilides. The extra lipophilic opioids are absorbed readily via the nasal or buccal mucosa. Opioids, particularly morphine, have been extensively used for spinal delivery to produce analgesia though a spinal action. These brokers show useful transdural movement adequate to allow their use epidurally. With most opioids, together with morphine, the impact of a given dose is much less after oral than after parenteral administration due to variable however significant first-pass metabolism in the liver. For example, the bioavailability of oral preparations of morphine is only about 25%. The form of the time-effect curve also varies with the route of administration, so the period of action usually is somewhat longer with the oral route. If adjustment is made for variability of first-pass metabolism and clearance, adequate aid of pain could be achieved with oral administration of morphine. Satisfactory analgesia in sufferers with most cancers is related to a broad range of steady-state concentrations of morphine in plasma (16�364 ng/mL) (Neumann et al. Compared with extra lipid-soluble opioids corresponding to codeine, heroin, and methadone, morphine crosses the blood-brain barrier at a considerably lower rate. About one-third of morphine in the plasma is protein certain after a therapeutic dose. The main pathway for the metabolism of morphine is conjugation with glucuronic acid. The two major metabolites shaped are morphine6-glucuronide and morphine-3-glucuronide. Although the 3-and 6-glucuronides are polar, both still can cross the blood-brain barrier to exert vital medical effects (Christrup, 1997). Morphine-6-glucuronide has pharmacological actions indistinguishable from those of morphine. Morphine-6-glucuronide given systemically is roughly twice as potent as morphine in animal models and in people (Osborne et al. Indeed, with chronic oral dosing, the blood ranges of morphine-6-glucuronide typically exceed these of morphine. In adults, the t1/2 of morphine is about 2 h; the t1/2 of morphine-6-glucuronide is considerably longer. In aged patients, decrease doses of morphine are beneficial primarily based on a smaller quantity of distribution and the general decline in renal operate within the elderly (Owens, et al, 1983). Morphine-3-glucuronide, one other important metabolite, has little affinity for opioid receptors however might contribute to excitatory effects of morphine (Smith, 2000). N-Demethylation of morphine to normorphine is a minor metabolic pathway in humans. Morphine is eradicated by glomerular filtration, primarily as morphine-3-glucuronide; 90% of the total excretion takes place during the first day. Enterohepatic circulation of morphine and its glucuronides happens, which accounts for the presence of small amounts of morphine in feces and urine for a quantity of days after the final dose. It shows a modest affinity for the receptor, but its analgesic actions are thought of by many to come up a minimal of partly by its hepatic metabolism to morphine (see further discussion). Thus, in distinction to morphine, codeine is about 60% as efficient orally as parenterally as an analgesic and as a respiratory depressant. Codeine is often employed for the management of cough, frequently together dose forms with acetaminophen or aspirin. Codeine analogues corresponding to levorphanol, oxycodone, and methadone have a high ratio of oral-to-parenteral efficiency. The larger oral efficacy of these medicine reflects decrease first-pass metabolism within the liver. A small fraction (~10%) of administered codeine is O-demethylated to morphine, and free and conjugated morphine can be discovered in the urine after therapeutic doses of codeine. The lowered sensitivity to morphine could also be because of decreased manufacturing of morphine-6-glucuronide (Caraco et al. Heroin is excreted mainly in the urine, largely as free and conjugated morphine (Rook et al. The drug is formulated in parenteral, rectal, subcutaneous, and oral preparations and as a nebulized formulation and is given off label by epidural or intrathecal routes. Hydromorphone has a better lipid solubility than morphine, leading to more fast onset than morphine, and is taken into account to be several occasions more potent than morphine. Oxycodone is available as single-ingredient medicine in immediate-release and controlled-release formulations. At current, oxycodone is among the mostly abused pharmaceutical medicine within the U. It is used orally for reduction of moderate-to-severe pain and is employed in a liquid formulation as a cough suppressant. It is approximately equipotent to oxycodone, with an onset of action of 10�30 min and length of 4�6 h. Oxymorphone is extensively metabolized in liver and excreted as the 3- and 6-glucuronides. These effects embrace respiratory despair, nausea, vomiting, dizziness, psychological clouding, dysphoria, pruritus, constipation, increased stress in the biliary tract, urinary retention, hypotension, and, not often, delirium. Increased sensitivity to ache may occur after analgesia has worn off, and removing of opiate receptor occupancy (abstinence, antagonism) may lead to a highly aversive state of withdrawal. In neonates or when the blood-brain barrier is compromised, lipophilic opioids may give more predictable scientific outcomes than morphine. However, because the pain subsides, the affected person may exhibit sedation and even respiratory despair because the stimulatory results of pain are diminished. All opioid analgesics are metabolized by the liver and must be used with warning in patients with hepatic illness. Renal disease also significantly alters the pharmacokinetics of morphine, codeine, dihydrocodeine, and meperidine. Although single doses of morphine are properly tolerated, the energetic metabolite, morphine-6-glucuronide, may accumulate with continued dosing, and signs of opioid overdose might outcome. This metabolite additionally could accumulate during repeated administration of codeine to sufferers with impaired renal operate. When repeated doses of meperidine are given to such sufferers, the buildup of normeperidine may trigger tremor and seizures. Similarly, the repeated administration of propoxyphene 372 � � � � � may lead to naloxone-insensitive cardiac toxicity caused by accumulation of the metabolite norpropoxyphene. There is a rising physique of information that examines gender variations within the responses to ache and analgesics (Mogil, 2012). Females have the vast majority of chronic ache syndromes, and surveys inspecting intercourse variations in acute pain models report both no sex distinction or larger sensitivity in females.
Azitrin 250 mg free shippingClorazepate is permitted as an adjunct remedy for the administration of focal seizures. Midazolam was designated an orphan drug in 2006 for intermittent therapy of bouts of elevated seizure 312 Plasma Drug Concentrations Because tolerance affects the relationship between drug focus and drug antiseizure impact, plasma concentrations of benzodiazepines are of limited value. It has relatively low toxicity, is cheap, and remains to be one of many more practical and broadly used antiseizure drugs. At levels exceeding therapeutic concentrations, phenobarbital additionally limits sustained repetitive firing; this will underlie a number of the antiseizure results of upper concentrations of phenobarbital achieved during remedy of status epilepticus. Therapeutic Uses Clonazepam is useful within the therapy of absence seizures in addition to myoclonic seizures in children. However, tolerance to its antiseizure effects often develops after 1�6 months of administration, after which some sufferers will now not reply to clonazepam at any dosage. The dose-dependent unwanted side effects are reduced if two or three divided doses are given each day. Clonazepam intranasal spray is designated as an orphan drug for recurrent acute repetitive seizures. While diazepam is an effective agent for remedy of status epilepticus, the efficient length of motion of this lipid soluble agent is shortened by its fast redistribution. Clorazepate is effective together with sure other medication within the treatment of focal seizures. In sufferers weighing more than 30 kg, clobazam is initiated orally at 5 mg every 12 h and then titrated as a lot as a maximum of forty mg/d if tolerated. It is 40%�60% bound to plasma proteins and bound to a similar extent in tissues, together with mind. The terminal t1/2 of phenobarbital varies widely, 50�140 h in adults and 40�70 h in children younger than 5 years of age, usually longer in neonates. Plasma Drug Concentrations Adverse Effects the principal side effects of long-term oral therapy with clonazepam are drowsiness and lethargy. Behavioral disturbances, especially in children, may be troublesome; these embrace aggression, hyperactivity, irritability, and difficulty in focus. Seizures are typically exacerbated, and standing epilepticus may be precipitated if the drug is discontinued abruptly. During long-term therapy in adults, the plasma concentration of phenobarbital averages 10 g/mL per every day dose of 1 mg/kg; in children, the worth is 5�7 g/mL per 1 mg/kg. The relationship between plasma focus of phenobarbital and antagonistic effects varies with the development of tolerance. Sedation, nystagmus, and ataxia normally are absent at concentrations below 30 g/mL during long-term remedy, but opposed effects may be obvious for a number of days at lower concentrations when remedy is initiated or whenever the dosage is increased. Concentrations more than 60 g/mL could also be associated with marked intoxication in the nontolerant individual. Because vital behavioral toxicity may be current regardless of the absence of overt signs of toxicity, the tendency to preserve sufferers, particularly children, on excessively excessive doses of phenobarbital must be resisted. The plasma phenobarbital concentration should be elevated above 30�40 g/mL provided that the increment is satisfactorily tolerated and only if it contributes significantly to management of seizures. Therapeutic Uses Antiseizure Barbiturates While most barbiturates have antiseizure properties, just some barbiturates, corresponding to phenobarbital, exert maximal antiseizure results at doses beneath people who trigger hypnosis. The pharmacology of the barbiturates as a class is described in Chapter 19; discussion on this chapter is restricted to phenobarbital and primidone. Phenobarbital is an effective agent for generalized tonic-clonic, focal-to-bilateral tonic-clonic, tonic-clonic of unknown onset (generalized tonic-clonic), and focal seizures. Its efficacy, low toxicity, and low price make it an important agent for these sorts of epilepsy. However, its sedative results and its tendency to disturb conduct in youngsters have lowered its use as a primary agent. Adverse Effects, Drug Interactions, and Toxicity Sedation, probably the most frequent undesired effect of phenobarbital, is clear to some extent in all patients on initiation of remedy, however tolerance develops during persistent medication. Phenobarbital can produce irritability and hyperactivity in kids and agitation and confusion in the aged. Scarlatiniform or morbilliform rash, possibly with different manifestations of drug allergy, occurs in 1%�2% of sufferers. As with phenytoin, megaloblastic anemia that responds to folate and osteomalacia that responds to high doses of vitamin D happen throughout persistent phenobarbital therapy of epilepsy. Concentrations of phenobarbital in plasma may be elevated by as a lot as 40% during concurrent administration of valproate. Primidone and its two metabolites each have antiseizure effects on focal and generalized tonic-clonic seizures. Primidone is completely absorbed and customarily reaches peak plasma concentration within about 3 h of oral administration. Both primidone and phenobarbital undergo extensive conjugation previous to excretion. In distinction, the terminal t1/2 of phenobarbital varies with age, with values ranging in adults from 50 to a hundred and forty h and in kids less than 5 years of age from forty to 70 h. Because of both gradual accumulation and clearance, phenobarbital reaches therapeutic concentrations roughly two to thrice larger than that of primidone. Like phenytoin, carbamazepine limits the repetitive firing of motion potentials evoked by a sustained depolarization of mouse spinal wire or cortical neurons maintained in vitro (McLean and Macdonald, 1986a). This appears to be mediated by slowing of the speed of restoration of voltage-activated Na+ channels from inactivation. The carbamazepine metabolite 10,11-epoxycarbamazepine also limits sustained repetitive firing at therapeutically relevant concentrations, suggesting that this metabolite may contribute to the antiseizure efficacy of carbamazepine. In addition to its early use in patients with focal-onset or generalized epilepsy, primidone is still considered to be a first-line therapy for important tremor with the blocker propranolol. Peak concentrations in plasma normally are observed 4�8 h after oral ingestion, but may be delayed by as a lot as 24 h, especially following the administration of a large dose. The predominant pathway of metabolism in people entails conversion to the ten,11-epoxide, a metabolite as active because the father or mother compound; its concentrations in plasma and brain could attain 50% of those of carbamazepine, especially in the course of the concurrent administration of phenytoin or phenobarbital. The 10,11-epoxide is metabolized further to inactive compounds which are excreted in the urine principally as glucuronides. Plasma Drug Concentrations Adverse Effects the dose-dependent opposed effects of primidone are much like these of phenobarbital, except that pronounced drowsiness is observed early after primidone administration. Common antagonistic results embody ataxia and vertigo, both of which diminish and will disappear with continued remedy. Clinical studies show the efficacy of felbamate in sufferers with poorly controlled focal and secondarily generalized seizures (Sachdeo et al. Interestingly, gabapentin additionally inhibits clonic seizures induced by pentylenetetrazol. Its efficacy in each of these checks parallels that of valproate and distinguishes it from phenytoin and carbamazepine. Rather, these compounds bind with high affinity to a protein in cortical membranes with an amino acid sequence similar to that of the Ca2+ channel subunit 2-1 (Gee et al. This interaction with the 2-1 protein might mediate the anticonvulsant results of gabapentin, but whether and the way the binding of gabapentin to the 2-1 subunit regulates neuronal excitability remains unclear.
References - Lipshultz LI, Corriere JN Jr: Progressive testicular atrophy in the varicocele patient, J Urol 117:175n176, 1977.
- Hautmann RE, de Petriconi RC, Volkmer BG: Lessons learned from 1,000 neobladders: the 90-day complication rate, J Urol 184:990n994, quiz 1235, 2010.
- Crivelli JJ, Xylinas E, Kluth LA, et al: Effect of smoking on outcomes of urothelial carcinoma: a systematic review of the literature, Eur Urol 65(4):742n754, 2014.
- Bergholz R, Koch B, Spieker T, et al: Polyorchidism: a case report and classification, J Pediatr Surg 42(11):1933n1935, 2007.
|
|


