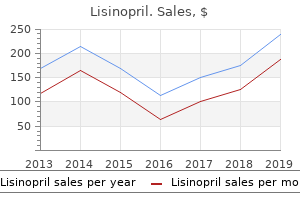
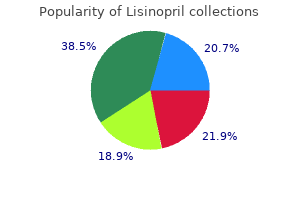
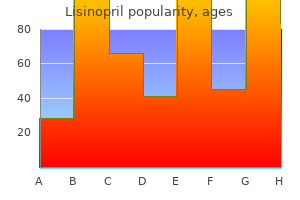
"Order lisinopril online, hypertension table in icd 9."By: Joshua C Briscoe, MD - Medical Instructor in the Department of Psychiatry and Behavioral Sciences
- Medical Instructor in the Department of Medicine
 https://medicine.duke.edu/faculty/joshua-c-briscoe-md
Buy lisinopril 2.5mg otcThe studies could be separated to think about (i) chemical and/or energetic stability, and (ii) microbial or bioburden management. Demonstration of microbial management is often performed at scale during process validation studies. The validated maintain time restrict will sometimes be outlined as the shortest time evaluated from three passing studies. These results are sufficient for supporting the method intermediate hold time limits. Filling: Agitation/Recirculation of Solutions Filling: Development Studies for Confirmation of Process Parameter Ranges and Targets the target of filling development research is to map design space parameters that produce constant delivery of the required quantity or weight of the compounded bulk drug product into the ultimate main container. Another objective of filling improvement studies, however, is to consider the impact of the filling process on drug product quality. For apparent reasons, these research require use of the active element of the formulated bulk. These studies ought to be carried out under protocol(s) and the results documented in improvement study final report(s). Establishing Limits for Operating Parameters Testing Approach Mixing research are needed for all new filling processes or main tools adjustments to existing processes. In precept, mixing on the filling line is comparable to mixing mentioned within the formulation course of (refer the "Mixing (Compounding)" section). Solution dosing vessel mixing occasions are finest studied with an on-line tank thermocouple, and results will range with agitator pace and coolant circulate rate via the tank jacket. Results from the study characterize tools capability and are necessary for informing preliminary process limits (min and max) on mixing velocity and time. Data Evaluation/Defining Operational Limits Acceptable results from the blending time course examine will outline the filling design house (the proven acceptable vary for operation). Establishing Limits for the Filling Line Start-up Parameters Once the transportable tank containing bulk product for filling has been related to the filling line, the agitator is began to ensure product homogeneity. Air in the filling line is purged from the tubing and needles through the preliminary dose setting. The initial dose is ready, the fill is began, and in-process management is implemented, or in-process dose checks are taken routinely throughout the fill (depending on type of filler being used). The initial line flush is based upon the calculated quantity of the filling line, and corresponding variety of pump strokes wanted to fill the line. Use the variety of calculated pump strokes required to fill the line as the begin line for variety of pump strokes to purge the road. An example calculation is offered below assuming the inner diameter of the line tubing is 1/8 in. Both the preliminary start-up and prolonged pauses (collectively referred to as "line downtime") typically end in materials which stays stagnant in some portion of the filling line. The impact is often because of settling (for suspension products), degradation (for temperature delicate products), and absorption into filling tubing (for small molecule products, preservatives, and/or small molecule components). In addition to these three factors, initial line start-up also requires consideration of complete priming of the filling strains. Different polymeric materials can have significantly completely different absorption traits. Initial start-up line flushing ought to use a purge as calculated above or the purge determined via the evaluation of downtime impression. Establishing Limits for Line Downtime Parameters the vital thing to efficiently managing downtimes during filling operations is to set up the dynamics of the stagnation time. Testing Approach There are usually three sets of experiments/evaluations required to fully characterize the process: (i) downtime dynamic characterization, (ii) hold-up quantity measurement, and (iii) flush-out dynamics. Downtime Characterization Laboratory-scale testing with mannequin tubing methods must be used to understand the time dependency of the vital thing parameter(s). For suspensions, you will want to think about the precise spatial orientation of the tubing when considering the settling dynamics. Since the move for this system during filling was basically plug move, the distribution of the alum was changed by the settling course of. Hold-Up Measurement For easy methods, the hold-up volume may be calculated directly. For instance, the calculation beneath assumes the inside diameter of the road tubing is 1/8 in. If direct measurement is used, the resulting elements can be used within the laboratory to decide flush-out dynamics. Flush-Out Dynamic the ultimate test required to set up the appropriate flush-out parameters is to consider the dynamics of the filling system. For simple systems, a flush-out quantity of 120% of the system hold-up quantity is recommended. The research ought to pattern product or a consultant surrogate after specified maintain occasions. It is typical to begin the sampling at 110% hold-up volume and continue to 200% hold-up volume. Evaluation of the downtime characterization knowledge and information of the manufacturing process must be used to choose the suitable downtimes to characterize. Data Evaluation/Defining Line Downtime Operational Limits Results from the downtime characterization studies are used to decide the allowed downtime prior to requiring a line flush. Evaluation of the impacted product characteristic(s) 2 Dose 3 Dose four Dose 2 Dose 1 Tubing Filling Needle Down Time (min) 5 10 20 Concetration of Alum (Target 0. Results from the flush-out dynamic study shall be used to determine the appropriate volume to flush. The appropriate quantity should then be transformed into variety of filled items based on the volume of fill. The strategy for setting the optimum VoF target for liquid vial merchandise may be separated into the next steps: � Establish the minimum fill requirement (inherent residual liquid on contact surfaces). Follow United States Pharmacopeia <1151> [3] steerage to establish the minimal necessities for vial overfill. However, the exact overfill must be adjusted based mostly on individual product traits. Conservative targets for VoF (excessive overage) end in financial loss because of wasted product remaining in the ultimate container after administration of the required dose, whereas aggressive targets for VoF (limited or minimal dose) may end up in prospects and/or patients unable to extract the label claim. The filling target depends upon the filling operation, assay variability, recoverable quantity from the ultimate container, and the product label claim. End-of-Fill Determination End-of-fill development studies are wanted to optimize the purpose at which the filling operation will be stopped during routine manufacturing. The goal of the research is to maximize crammed container yield while ensuring product high quality (consistency of dose and content).
Amorphophallus konjac (Glucomannan). Lisinopril. - Helping control type 2 diabetes.
- What other names is Glucomannan known by?
- Constipation, weight loss, and high cholesterol when used with chitosan.
- Are there safety concerns?
- Dosing considerations for Glucomannan.
- Are there any interactions with medications?
- What is Glucomannan?
- How does Glucomannan work?
Source: http://www.rxlist.com/script/main/art.asp?articlekey=96238
Best order lisinoprilProgression of alcoholic and non-alcoholic steatohepatitis: common metabolic features of innate immune system and oxidative stress. Noninvasive prediction of clinically important portal hypertension and esophageal varices in patients with compensated liver cirrhosis. Hepatic encephalopathy in continual liver illness: 2014 practice guideline by the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver. Choose acceptable pancreatic enzyme supplementation for patients with persistent pancreatitis. Many pseudocysts resolve spontaneously, but some require surgical or percutaneous drainage. The exocrine cells of the pancreas are referred to as acinar cells that produce and retailer digestive enzymes that mix with a bicarbonate-rich resolution launched from duct cells to produce pancreatic juice. Amylase and lipase are released from the zymogen granules in the lively kind, whereas the proteolytic enzymes are activated in the duodenum by enterokinase. Enterokinase triggers the conversion of trypsinogen to the lively protease trypsin, which then activates the other proenzymes to their energetic enzymes. Fluid necessities should be assessed fastidiously, particularly in patients with concomitant cardiac, renal, and liver disease. Electrolytes corresponding to potassium and magnesium may be added to the infusions if necessary. Early use of enteral vitamin (within forty eight hours)10 has been proven to decrease surgical interventions and infectious complications. In necrotizing pancreatitis, antibiotics could additionally be acceptable for sufferers who fail to enhance after 1 week, or deteriorate. The decision to use antibiotics ought to be guided by fantastic needle aspiration whenever attainable. Stable patients with infected necrosis must be treated with antibiotics; surgical, radiologic, and/or endoscopic drainage would ideally be delayed by four weeks to allow for the area of necrosis to become walled off. Infections are often polymicrobial, so broad-spectrum antibiotics with activity towards enteric gram-negative bacilli are appropriate (Table 23�2). Patients might receive long courses of broad-spectrum antibiotics and may develop superinfections with resistant bacteria. What information about the patient presentation is in maintaining with acute pancreatitis Patient Care Process for Acute Pancreatitis Collect Information: � Assess fluid and dietary standing and belly pain severity utilizing a pain scale. Assess the Information: � Review laboratory information, imaging and medicine historical past for potential threat elements for pancreatitis (Table 23�1). Ineffective Therapies Therapies with no proven benefit on morbidity and mortality embody reducing pancreatic secretion by administering somatostatin analogues or atropine, decreasing gastric acidity and pancreatic secretion with histamine2-receptor antagonists, probiotics, and immunomodulation. Symptoms � Pain is the most common symptom, and it usually starts in the epigastrium and may radiate to the back or scapula. Pain can be episodic with pain-free intervals, or extended and requiring hospitalization. One concept is that protein-rich plugs kind resulting in ductal obstruction and inflammation, ultimately inflicting parenchymal fibrosis and ischemic harm to the acinar cells. As pancreatic exocrine function diminishes, sufferers have decreased capacity to take up lipids and protein with regular dietary consumption, leading to weight loss and malnutrition. Fat- or proteincontaining stools are widespread; carbohydrate absorption is normally unaffected. Patient Encounter 1, Part 3 the patient was discharged and presents again to the hospital 3 weeks later with similar pain and feeding intolerance. Besides antibiotics, what different pharmacologic therapies may need to be optimized Alcohol and cigarette abstainers might have slower disease development and better response to pain remedy than nonabstainers. Indications for surgical procedure embody poorly controlled ache, ductal obstruction, and symptomatic pseudocysts. Patients can require persistent doses of opioid analgesics, with resulting threat of dependancy. He has also had light-colored liquid stools for the previous month and has misplaced 10 kilos (4. The half-life of endogenous lipase is predicated on the presence of its substrates (ie, triglycerides), and therefore dietary fats restriction ought to be reconsidered when pancreatic enzyme substitute is used. What, if any, therapy regimens would you initiate, and the way would you monitor their results Assess Information: � Determine whether or not the affected person is receiving any medication(s) that will trigger or exacerbate pancreatitis. Implement the Care Plan: � Provide sources for alcohol and smoking abstinence, if relevant. L O 4 surroundings and due to this fact have to be administered with a proton pump inhibitor. The traditional starting dose is 500 units/kg/meal of lipase with half of the mealtime dose administered with snacks. Doses of lipase greater than 10,000 units/kg/day must be used with caution as a outcome of they may be related to colonic stricture and fibrosis. Fasting blood glucose should be monitored annually, and ensuing diabetes must be handled with insulin. Chronic pancreatitis: challenges and advances in pathogenesis, genetics, analysis and remedy. The prevalence of malnutrition and fat-soluble vitamin deficiencies in chronic pancreatitis. The prevalence of fatsoluble vitamin deficiencies and a decreased bone mass in sufferers with persistent pancreatitis. Pregabalin reduces pain in patients with continual pancreatitis in a randomized, controlled trial. Long-term prognosis of autoimmune pancreatitis with and without corticosteroid therapy. A randomized controlled trial of antioxidant supplementation for pain reduction in sufferers with continual pancreatitis. Antioxidant therapy for sufferers with persistent pancreatitis: a scientific evaluation and meta-analysis. An worldwide multicenter research of early intravenous fluid administration and consequence in acute pancreatitis. Differentiate the five types of viral hepatitis by epidemiology, etiology, pathophysiology, clinical presentation, and natural history. Identify modes of transmission and threat components among the major kinds of viral hepatitis. Formulate a monitoring plan to assess opposed effects of pharmacotherapy for viral hepatitis. Acute hepatitis may be related to all five kinds of hepatitis and barely exceeds 6 months in duration.
Order lisinopril onlineIn this process, the polymer is extruded by way of a multi-hole die, and the polymer stream is stretched and attenuated by a high-velocity heated airstream. The imply fiber diameter is changed as the filter is being made by adjusting the air velocity or one of the different variables that contribute to the formation of the fiber sizes, for example, temperature or polymer pumping price. This technology is turning into extra advanced, with some producers naming the fibrous fleece constructions as nano-fiber fleeces. The concept of utilizing a graded or changing pore size to improve filtration efficiency is a desirable one. This method entails incorporating a series of prefilters into a single stage to maximize the use of the entire filter and lengthen filter life (dirtholding capacity). The factor of fractionate retention is especially important for functions with a wide particulate spectrum, for example, water pretreatment. Prefilters can even contain membranes, porous, or fibrous, generally from cellulose, mixesters, or borosilicate. These prefilter types are utilized to remove a really nice band of particulate or contaminants from the fluid to particularly defend sterilizinggrade membrane filters. Highly adsorptive cellulosic or kieselgur containing depth filter pads are welded together in a plate format. These plate codecs generally have a diameter of 12 or sixteen and comprise stacks of 4�16 to create a depth filter unit. The adsorptive depth filter materials is ideal to separate colloidal substances and lipids; as a result, these filters were fairly often utilized in plasma and serum applications. Nowadays, lenticular filters are most often utilized in cell harvest purposes after the fermentation course of to separate cell debris and other contaminants. When lenticular filter combinations are examined, the checks not only involve the total throughput of the filter factor as is commonly the case with pleated prefilter cartridges, but an important issue is the turbidity measurement of the filtrate. The turbidity measurement will create an indication of the protecting properties of the lenticular filter retention score used and how a lot of the contaminants are separated by the actual filter ranking. Small-scale checks are carried out to determine which filter mixture is ideal and meets the process necessities. The report also addresses the problem of the release of beta glucans from some of the depth filter materials, which might create a false failure within the endotoxin take a look at ranges. The filters require thorough flushing to avoid any undesired leaching into the product stream. Prefilters Prefilters are mostly depth filter varieties and are typically constructed of nonwoven or melt-blown fiber materials similar to polypropylene, polyamide, cellulosic, glass fiber, metal fibers, and sintered stainless steel (12). Normally, prefilter supplies are constructed into mats by the random deposition of either individual or steady fibers whose fixation is accomplished by urgent, heating, gluing, entanglements, or other varieties. The pores of such filter constructions are rather random interstices among the fibers. The casting solution consists of polymer dissolved in a combination of solvent and high-boiling non-solvent. Pore formation happens as follows: as solvents progressively evaporate from the casting solution, the non-solvent increases in content material to the purpose the place section separation takes place. Non-solvent droplets separate within the polymer/solvent section, and polymer comes out of solution to focus on the droplet interfaces. The swollen polymer shells surrounding the non-solvent droplets thicken as continuing solvent loss causes extra polymer deposition. The eventual disappearance of the polymer/solvent phase brings the polymer-surrounded droplets into mutual contact. They consolidate into clusters and deform into polyhedral cells full of nonsolvent underneath the impetus of the world minimizing forces. Finally, the perimeters of the cells accumulate polymer on the expense of the cell walls. Thinning of the walls of the polyhedra results in their rupture and interconnection. The reticulation of the discrete cells of the polymeric matrix permits the removal of the non-solvent, as by washing. Not the polyhedral cells, however their interconnecting openings, thus formed, comprise the metering pores of the membrane (15). Nanofilters Most generally, nanofilters are designed to separate viruses, utilizing measurement exclusion as the predominate mechanism of removal. Since nanofilters are extremely tight filters, the water bubble level is commonly larger than the maximum allowable operating stress. Therefore, integrity testing of those filters requires special take a look at methodologies, corresponding to liquid porosimetry (13,14). This test technique makes use of two immiscible liquids which are successively intruded by strain into the biggest pores of the membrane. These porosimetry measurements may be correlated to viral removal post-filtration, which allows the check to be used to validate viral elimination in precise practice. Nanofilters or viralretentive filters are an essential contaminant elimination step, particularly in bioprocesses. A multitude of nanofilters can be found for different applications and target contaminants. Most common retention ratings are 20 and 50 nm, also identified to separate parvo and retro viruses. The oversimplified image of the filter pores is that of irregular and tortuous capillaries composed of the interconnected spaces inside the polymer matrix. The construction derives from a polymer resolution, and distances that replicate the polymer dilution separate the chain segments from one another. It is the ultimate interstitial distances that, of their interconnections, prefigure the pores of the finished membrane. Formulae of different Polymeric Types and Properties As one can count on, there are distinct variations between the person membrane and prefilter polymers. Therefore, filter membranes and filter efficiency have to be examined earlier than selecting the appropriate filter component. Membrane filters have a very low leachable level and therefore are used at critical filtration steps, for example, before filling. Some membranes are cost modified; these filters have a special surface remedy on the membrane to create a highly optimistic or adverse cost for particular contamination elimination purposes. Pores Size Ratings Where sieve retention of particles is the one consideration, the scale of the biggest pore current in the filter is of ultimate concern. Sieve retention is consequently assumed to be the sole particle-capture mechanism operational. This is the supposed scenario, for the dependability of sieve retention is seen in its freedom from the operational components that influence the efficiencies of adsorptive removals, such as the organism challenge level, the magnitude of the applied differential strain, and even such parameters as fluid temperature, viscosity, ionic strengths, and the presence of wetting brokers that represent the contribution of the liquid car (16,17).
Discount lisinopril online mastercardTo gain optimum circulate rates from membrane filters, there are limited parameters, which may be managed within the filtration course of. Either the differential stress can be raised or the bigger filter surface areas may be applied, with the disadvantage of increases in consumable, unspecific adsorption, holdup quantity, and capital investment costs. The circulate price optimization of filtration processes requires exams using comparable filter parts, commonly 10 filter cartridges. The test could be performed under the specified course of conditions, commonly using a set inlet pressure, whereas the time to filter the fastened fluid quantity is measured. It is important that the process parameters are kept constant, that means the same buffer composition, pressure, and temperature settings must be applied. Total Throughput Total throughput, that means the total quantity filtered before the filter factor blocks, might be probably the most extensively required efficiency standards in most filtrative applications. It is directly proportional to the filter design, surface area, system measurement, and prefilter mixtures. Total throughput has a serious impact on filtration prices, and what would possibly appear to be a cheaper filter may very well considerably enhance the filtration costs. The total throughput of a filter cartridge is dependent upon the membrane filter polymer, pore construction, and filter design. Some membrane polymers are adsorptive, and better adsorptivity is often associated with a higher fouling price and therefore lower complete throughput. A coarser prefilter membrane layer in front of the ultimate filter membrane, the so-called heterogeneous double-layer membrane, has a distinctly greater whole throughput. It can be expressed as the mass of particulate matter held by a filter or by the amount of fluid supplied to a filter, assuming that the filth concentration of the suspension is homogenous and fixed. The capacity of a filter is a measure of the entire quantity of fluid that might be processed earlier than a pressure drop develops to an unacceptable stage. The objective is for an entire and timely processing of a manufacturing run, well timed being defined by means of sensible and financial significance as nicely as retentivity assurance (7,8). Throughputs may be judged inadequate if, given the chosen circumstances of accessible filter area and differential strain, the quantity of effluent produced is at too gradual a rate to meet the time necessities of the operation. If the filtration is already underway, increasing the filter area is the harder various. Raising the differential stress, though easier to accomplish, deserves considered utility. The whole throughput can be further advanced by evaluations of applicable pre- and last filter combos, if required. The differential stress, temperature, and product conditions stay the identical in all trial cases. The theoretical instance depicts that a lower price prefilter might be used to defend the ultimate filter and scale back the required ultimate filter dimension. Total throughput tests to determine the appropriate ultimate filter and/or mixture of pre- and final filter are carried out with forty seven mm flat filter composites. These composites have to have the same fleece and filter mixture because the filter component to be used later. Commonly, multiple composites are examined to determine the suitable ultimate filter and to be succesful of take a look at multiple prefilter options. To define the right filter size required inside the production course of, small-scale pleated gadgets of the predetermined filter combination must be utilized. Unspecific Adsorption Unspecific adsorption on filter membranes is another leading cause of yield loss within biopharmaceutical processes including protein degradation due to proteolysis. Any yield loss is proportional to the loss of manufacturing capability and market value. Therefore, unspecific adsorption testing have to be a precedence within applications that could be adsorption delicate. Applications encompassing drug merchandise containing preservatives and therapeutic proteins are frequent examples of adsorption-sensitive processes. Protein surfaces can contain completely different hydrophobicity, charge, and degree of hydration, and might change with protein conformation and with solution traits (9). Therefore, hydrophobic adsorptions are believed to mirror protein�filter interactions. Shifts in round dichroism and reduces in enzyme exercise resulted from conformational changes of the protein construction. Protein� membrane interaction triggered the protein to expose its hydrophobic sites, which have been folded within its structure throughout its exposure to aqueous solution. As described in testing for complete throughput, commonly, unspecific adsorption assays are carried out throughout small-scale trials or inside the course of validation procedures of a filter into the precise product and process specifications. Small-scale trials ought to be carried out as early as potential so as to avoid any surprises or possible validation delays additional down the event course of. As these trials generally make the most of a small quantity of the particular drug product, optimization trials can additionally be performed. For example, in certain applications, it has been discovered that buffer flush, specific pH, or temperature circumstances can minimize the unspecific adsorption onto the filter membrane. These circumstances require evaluation besides the precise membrane filter polymer and composite. The larger the area or the extra membrane layers which are utilized, the higher the adsorption. Changes within the described circumstances and the casting solution combine will create completely different pore structures, porosities, and membrane structures. In the quenching course of, the polymer/solvent combine is utilized onto a drum or belt, which immerses into a solvent or extraction bathtub. Melt extruded movies are stretched beneath defined course of conditions to create a skinny membrane. The thinnest (10�20 �m) membrane movies are created by track-etched manufacturing course of. Types of Filters Membrane Filters Membrane filters generally have a defined pore construction and porosity band. The narrower the porosity band, the more outlined the retention fee of such membrane is. The filtration obtained by the use of such membrane filters is commonly referred to as microfiltration. Microporous membrane filters have a method more defined porosity than is available inside prefilter matrixes. Membranes are produced by either an evaporation, quenching, stretching, or track-etched process. The pore structure of track-etched membranes may be very defined, however due to the avoidance of particle observe overlaps, the porosity is low. Membrane filters could be shaped in a variety of constructions for particular software functions.
Cheap lisinopril master cardThe pointers suggest that the frequency of microbiological monitoring be primarily based on the sort of processing actions and the time taken for processing and be enough to permit for effective monitoring of potential microbiological contamination of sterile pharmaceutical merchandise. Manufacture of sterile merchandise requires managed environmental circumstances and monitoring of particulate and microbial ranges in the manufacturing areas. Particles are important as a end result of they may enter a product and contaminate it physically or by appearing as a automobile for microorganisms. Air supply systems are characterized by particular parameters (temperature, humidity, air pressure, air adjustments per hour, velocity, and so forth. Microbiological contamination and monitoring are critical parts in sterile manufacturing. Microorganisms in the air are typically in combination with particles; nevertheless, it is essential to note that there is probably not a direct correlation between the number of particles within the air and microbiological contamination. Therefore, microbiological testing is a vital measure of assuring high quality and security of products produced under sterile circumstances. Standard practices related to environmental monitoring may differ between producers by way of the frequency, variety of sampling sites, methods and tools utilized in sampling, etc. However, it may be very important keep the validated state and have procedures in place to document and remediate any deviations from the validated state. Criteria for "most allowable particles" are the same for each aseptic and terminal sterilization processes. For initial qualification, all tips specify that course of simulation be performed using three media fills and as acceptable, include all personnel shifts in the simulation program. Media fills should reflect routine commercial production and embody worst-case actions and circumstances that challenge aseptic operations in order to precisely assess the potential for contamination. Process simulations must additionally tackle all dangers to sterility assurance specific to the person processes. Measurement time per plate is 4 h at maximum, and the measurement is carried out during processing operation. These developments are then used to establish out-of-trend information and occasions, provoke root cause investigations, and take corrective actions. There are, nonetheless, minor differences in expectations and focus areas for trending of environmental knowledge. Isolator Technology and Blow/Fill/Seal Aseptic processing utilizing isolation systems separates the exterior cleanroom surroundings from the aseptic processing line and minimizes its exposure to personnel. A well-designed positive pressure isolator, supported by sufficient procedures for its upkeep, monitoring, and management, presents tangible benefits over conventional aseptic processing, including fewer opportunities for microbial contamination during processing. This manufacturing expertise contains economies in container/closure processing and lowered human intervention and is commonly used for filling and packaging ophthalmics, respiratory care products, and less regularly, injectable products. Other steerage paperwork handle these matters however in various level of detail and cover features such as validation requirements and environmental monitoring. Drug product parts, containers, closures, storage time limitations, and manufacturing equipment are among the many areas to tackle in establishing endotoxin management. Endotoxin testing is required by international well being authorities, and acceptance standards have to be specified within the regulatory dossier. In the event of doubt or dispute, the gel-clot restrict test should be used to make the ultimate determination on compliance for the product being examined. Therefore, the manufacture might need to assess each proposed change and decide the danger associated with the change. The danger assessment should be designed to adequately assess the effect of the change on the drug substance and drug product. The following are examples of factors to contemplate when conducting a threat evaluation associated to a change: � Experience of the manufacturing facility and/or personnel involved within the portion of the process that encompasses the proposed change � Changes carried out at the same facility that has adequate expertise with the type of change versus a brand new facility with inadequate experience. The report supplies threat assessments for lots of the most typical forms of manufacturing changes and can be used as an excellent reference doc whereas making risk-based regulatory choices including regulatory influence assessments for post-approval changes. To consider the danger, frequency (F) which describes the chance of prevalence, severity (S) or consequence from the event, and the extent of detectability (D) had been evaluated for every occasion utilizing the danger evaluation scale in Table 49. The level of danger is then determined as follows: Risk = (S) � (F) � (D) Recommended reporting classes upon completion of the risk assessment are offered in Table 49. Some basic assumptions will need to be made and factored into the danger assessments. Minor: Has restricted potential to have an opposed effect on high quality and/or safety of the drug. Moderate: Has a moderate potential to have an antagonistic impact on quality and/or safety of the drug. Major: Has a considerable potential to have an adverse effect high quality and/or security of the drug. Case Study 1: Facility-Related Change- Change in Area Classification Description of the Change: Change in space classification (decrease in class/grade) within the non-filling space for aseptic processing where the manufacturing process is open (product exposed) and the product stream does 1076 not support microbial growth. Bioburden increases within the product prior to the filtration step exceeding the retentive capabilities of the sterilizing filter and resulting in a non-sterile product. Bioburden will increase in the product previous to sterilization inflicting an increase within the endotoxin levels in the product. Parenteral Medications Overall threat level: 12 Submission Category: Based on the level of danger and the suggestions for filing categorization in Table 49. Case Study 2: Facility-Related Change-Change in Site for Aseptically Processed Product Description of the Change: Change in web site for an aseptically processed product � Same campus/same building/new or refurbished area line � Same campus/different building/new or refurbished space or line � Different site/same company/new or refurbished area or line � Different site/different company/new or refurbished area or line. Decreased level of sterility assurance ensuing within the production of non-sterile product. Increased variability of filling resulting in product with inaccurate fill volume/dose. Major: Has a considerable potential to have an antagonistic effect high quality and/or security of the drug (4). Increase in the variety of filters (and subsequently, product exposure to the filter components) leads to a reduction of drug product potency due to increased product absorption. Increase within the number of filters (and due to this fact, product exposure to the filter components) results in a rise within the degree of extractables/leachables in the drug product. Overall danger stage: eight Submission Category: Based on the level of threat and the suggestions for submitting categorization in Table 49. It is anticipated that rapid development within the field of gene and cell therapy merchandise, that are invariably parenteral injectables, will result in frequent new (or updates of current) guidance. Global Sterile Manufacturing Regulatory Guidance Comparison: Parenteral Drug Association Publication, 2016. As consultants, our clients often ask us what the future of parenteral manufacturing might be. First, they will need to have potential relevance within the subsequent decade and beyond; and second, no much less than certainly one of us was educated enough to make a meaningful effort to predict what the lengthy run circumstances might be.

Buy lisinopril 10 mg overnight deliveryOver the intervening 50 years, radiation in the type of highenergy electrons produced by high-power accelerators or gamma rays produced by radioisotopes has been used to terminally sterilize a broad spectrum of medical devices and various varieties of pharmaceutical merchandise. More just lately, a 3rd supply of radiation, X-ray (Bremsstrahlung) radiation, has been introduced to the radiation sterilization business. Because radiation sterilization is classified as a chilly course of, it may even be used to sterilize heat-labile merchandise. This article begins with a discussion of the fundamentals related to the interaction of radiation with materials. A part is devoted to dosimetry, a critical a part of the process, which varieties the premise for dosimetric launch of product. All three modalities which may be presently used for radiation sterilization of merchandise are lined, including design and operation of irradiators and control of the irradiation setting. The last sections of the chapter handle radiation chemistry of liquids and solids, radiation effects, and a last section on irradiation of specific drug merchandise. Interaction of Radiation with Material Sources of Radiation Three sources of radiation are used in the radiation sterilization process. Gamma rays are pure electromagnetic power within the type of quanta of radiant power called photons. The primary isotope used in the radiation sterilization industry is cobalt-60, which emits two photons per disintegration of the nucleus with energies of 1. Another isotope, rarely used because of its solubility, is cesium-137, which emits one photon per disintegration at an vitality of 0. Cobalt-60 is the isotope of choice for business functions of radiation processing. It can be manufactured in a metallic form, which is inherently secure and produced in much larger particular actions (~100 Ci/g) than cesium-137. Because of its high particular activity, cobalt-60 may be fabricated in compact energy-efficient sources. To obtain the megacurie levels of activity that are used in commercial gamma irradiators, literally hundreds of these sources are used to construct the supply plane(s) in a commercial irradiator. Its half-life, which is a measure of the speed of decay of the radioactive isotope, is for much longer than that of cobalt-60. The halflife of cesium-137 favors its use in self-contained irradiators corresponding to blood irradiators which are used to prevent transfusion-induced graft-versus-host disease. However, cesium-137 is water soluble and is subsequently not licensed for use in commercial irradiators in the United States or most different nations. The second supply of radiation consists of high-power accelerators that generate high-energy electrons. Depending on the accelerator design, electron energies can vary from lower than 1 MeV to over 10 MeV. The third source of radiation happens when the high-energy electrons from a high-power accelerator impinge on a high atomic weight conversion target. Because the conversion effectivity increases because the sq. of the atomic variety of the goal material, conversion targets are fabricated from high atomic number materials. The high-energy electrons that impinge on the conversion goal are deflected within the area of the nucleus of the atoms within the conversion goal and in the process of changing (stabilizing) orbitals, emit electromagnetic radiation in the form of X-rays. This type of electromagnetic radiation is referred to as Bremsstrahlung radiation, which translated means "braking radiation. A widespread attribute of the three sources of radiation which are employed within the radiation sterilization process resides in the truth that the power of the incident radiation is adequate to ionize the atoms that make up the molecules of the materials (and/or bioburden) which would possibly be irradiated. In the ionization process, enough power is imparted to the orbital electrons of an atom to take away the electrons from their sure state around the atom. Depending on the specific factor, the vitality wanted to remove the outermost electrons. Because the power of the incident radiation is measured in MeV or millions of eV, adequate energy is obviously obtainable to initiate the cascading ionization course of. For this purpose, these radiations are referred to as ionizing radiations and are differentiated from nonionizing radiation corresponding to optical radiation. The energy of the photons from these sources of electromagnetic radiation is lower than a couple of eV, which is insufficient to ionize an atom. The power loss per collision is relatively low in comparability with the energy of the incident electron. For instance, the average energy loss per scattering occasion for a ten MeV electron is roughly a hundred eV per collision or less [3]. It is of interest to observe that the power misplaced by primary electrons which might be produced by Compton scattering of photons happens in the identical manner. Therefore, regardless of the modality of the incident radiation, the vitality switch mechanisms that result in the sterilization of medical devices/pharmaceuticals and changes in material properties are the same. The results of high-energy photons are indistinguishable from those produced by the identical amount of vitality per unit mass (absorbed dose) imparted by high-energy electrons. This equivalence of effects is the basis for the utilization of electron beam, gamma, or X-ray forms of radiation in radiation processing. However, the rate of energy deposition for the completely different sources of radiation may be fairly completely different, and this parameter may play an necessary position in the resultant effect on supplies which would possibly be irradiated. Interaction of High-Energy Photons with Materials At intermediate photon energies that characterize the gamma ray and X-ray (Bremsstrahlung) sources of radiation used in the radiation sterilization course of, the dominant channel for interaction of the photons with the orbital electrons occurs through a course of called Compton inelastic scattering. This methodology of vitality switch is called after the individual who first described the quantum mechanical relationships governing the scattering course of [2]. A photon undergoing Compton scattering transfers part of its energy to the orbital electron. The quantity of vitality transferred to the electron will depend on the quantum mechanical relationships governing the scattering event however is often enough to not solely ionize the atom but in addition go away the electron with important kinetic energy to continue the ionization course of. In truth, probably the most possible Compton scattering occasion is a backscatter of the photon, which transfers maximum power to the electron. For gamma rays emitted by a cobalt-60 supply, a backscattered photon will deliver about 1 MeV to the orbital electron. The scattered photon continues to endure scattering occasions and generate extra main electrons until its vitality is dissipated. The main electrons have adequate energy to ionize different atoms through an electron�electron inelastic scattering process. The photon features as an initiator of the method that leads to radiation sterilization throughout the majority of the medical device/drug product. Energy from the incident radiation is transferred to the fabric by varied pathways as discussed in the earlier section. It is defined as the quantity of ionizing radiation power imparted per unit mass of a specified material [4]. The previous unit that was used to measure absorbed dose was the rad, which is not in use (100 rads = 1 Gy).
Syndromes - Blood chemistry tests (such as albumin level)
- Blood pressure changes -- may be high or low
- Alcohol use
- Cigarette smoking
- Coma
- Vitamin K deficiency
Buy lisinopril visaThe validation manager inadvertently met the manufacturing manager and asked in regards to the cleaning. The production supervisor proudly stated that the equipment had been cleaned by their greatest operator and that the equipment was cleaned by the operator thrice. Production senior management had skilled a quantity of current cleaning validation failures and informed their workers to do no matter was necessary to cross cleansing validation. Operators should understand that cleaning procedures are equivalent to manufacturing procedures. Operators have to be adequately educated in efficiency of the manual cleansing process. Training on cleaning procedures requires actual efficiency underneath the supervision of area specialists. The final step in the cleaning procedure after equipment is dry is the inspection of the cleaned equipment. Operators ought to pay consideration to components that may cause erroneous inspections, similar to inadequate mild and equipment wetness. All personnel, including quality unit personnel involved with gear inspection, also needs to have coaching in correct inspection strategies. Maintaining and monitoring handbook cleaning performance: Operators who perform handbook cleansing procedures have to be periodically noticed to maintain the validated state of the cleansing course of. Periodic evaluation insures that the operator efficiency is in keeping with written procedures, which in turn are consistent with the original cleansing validation. At the identical time, improvements in the cleansing procedure may be identified during the evaluation. Manual cleaning is a high-risk activity because of the inherent variability of personnel efficiency. As such, it should be rigorously designed and developed, carried out as said in procedures, and continually monitored. Carefully written procedures, including specific details of important parameters, are an important side of guide cleansing procedures. Key parts to guarantee good guide cleansing procedures include good technical development, clearly written procedures with specified process parameters that are neither too detailed nor too vague, performance by well-trained operators, and interval monitoring to keep validation. Awareness of these key components by manufacturing personnel is crucial to the success of handbook cleansing procedures. Cleaning validation practitioners should be confident that persons doing handbook cleansing are properly skilled. Monitoring and upkeep of handbook cleansing processes should be commensurate with process threat. Regulatory evaluation of manual cleansing processes: Manual cleaning is an space of recurring deficiencies during regulatory inspections. More often than not, guide cleansing processes are too sparsely proceduralized by way of the steps which might be wanted to carry out the cleansing. This introduces the potential for broad variability in the actual cleansing process used, both between Manual Cleaning Personnel Qualification Manual cleansing is cleaning done completely or partly by humans. The dimension, energy, and handbook abilities of individuals contribute to potential variation in manual cleansing processes. Manual cleaning procedures could comprise the whole strategy of cleansing manufacturing equipment or could also be carried out along side automated cleaning methods. Manual cleaning procedures should be considered as equal to handbook manufacturing procedures regarding total technique and method. Any procedures that have a significant human component should observe a lifecycle strategy to validation. They have to be appropriately developed, validated, and maintained with the inherent manual variation in mind. The following are key issues for efficient manual cleansing procedures: Stage 1. Manual cleansing course of growth: Manual cleansing procedures must be developed with considerations for human variation. Technical parameters must be determined considering the inherent variability of manual cleansing procedures. Performance by totally different manufacturing personnel-with completely different levels of bodily dimension, power, and private motivation, should be thought-about. Performance on first, second, and third shifts ought to be anticipated to be totally different. Manual cleaning efficiency: Operators who might be doing the precise work of cleaning should have a basic information of the cleaning procedure being carried out. Operators who carry out the cleaning process must clearly understand that the main points of the process are inviolate-not something like washing dishes at home. Operators should now perceive that procedures must be explicitly carried out within the order specified within the cleansing procedure. In addition, the data that are made up of manual cleaning operations are often not sufficiently detailed. Some corporations use a check-sheet approach by which just one check is made to doc that a multistep cleansing course of has been performed. In such cases, no record may be made up of what, if any, detergent was used, whether hot or chilly water was used, or how long the cleaning process took. There are arguments supporting this belief-but there are also arguments supporting the other. Whether the term "validation" or another term is used in relation to cleaning processes which are used to clean non-dedicated product-contact tools, the key concern is that the method must be shown to be efficient and fit for its meant use, that the equipment can be cleaned to the required diploma every time, and that the extent of variability is minimized. In working to achieve this, the nature of the gear to be cleaned must be thought-about, as nicely as the services which are available for handbook cleaning activities. The latter can have metal components in addition to plastic and soft components, such because the flexible Teflon chutes that may feed the fabric into drums, and the cleaning strategies for each of those components must be thought-about. Parenteral Medications allow variation in the cleaning process-the antithesis of a validated process. Cleaning procedures must be documented in the same manner as manufacturing procedures-in the format of batch information. The performance of key steps within the cleansing procedure, simply as in manufacturing procedures, must be affirmed by operator signoff. Note operator signature/data really helpful for particular person important steps solely; noncritical step could additionally be grouped for single signature/date. Manual cleansing procedures with specified important parameters requiring operator signature/date are particularly necessary. Minimizing cleansing process variation can only be achieved with rigorously written procedures. For instance, in a washroom used to manually clear small gadgets of kit, one ought to check that the cleaning process specifies the place cleaned gear left to dry is to be stored-the danger of contaminating already-cleaned equipment when other objects of equipment are present process cleaning have to be carefully managed.

Purchase lisinopril 5mg amexThe manufacturing methods had been bigger and more subtle, and the residues extra diverse, so whereas the design of the disposable unit is more advanced (and expensive), this was more than offset by the elimination of the difficult and prolonged cleansing process and cleansing validation. Potential purposes for disposable techniques in parenteral manufacturing and filling are limited only by the creativeness of the end person. Small-scale filling units, process filtration assemblies, aseptic sampling systems, and other gadgets are broadly available as disposable assemblies, which could be both a "normal" configuration or customized for a selected utility. The increased use of disposable techniques is definite given the operational advantages to their use, because the benefits prolong well past eliminating cleaning and cleaning validation. The smaller disposable systems and assemblies provide substantial advantages together with increased certainty of assembly; lowered labor prices for system preparation, cleansing and sterilization; lowered needs for internal sterilization equipment and process time; enhanced confidence in sterilization; and higher system integrity. The polymeric supplies utilized must not work together with the materials being processed in any way. The potential for extractable and leachable materials to work together with the product should be evaluated to guarantee that no adverse effects are current. Similarly, the adsorption/ adherence of formulation components by the polymeric materials should even be thought of. The issues here are corresponding to these related to the utilization of membrane filters; nonetheless, the exposure periods are typically longer and the conditions of Parenteral Medications use more variable. Discarding costly and enormous polymer containers and tubing after a single use runs counter to the increasing importance of the environmental safety. It appears logical that single-use expertise vendors will consider means by which their merchandise could be recovered and recycled. The use of disposables techniques offers sufficient advantages that increased use sooner or later is assured. Filter Pore Size Filtration has been used to take away microorganisms from parenteral and ophthalmic solutions for many years. During the primary half of the last century, many sterilizing filters had been made of asbestos or unglazed porcelain supplies and achieved bacterial retention mainly by way of depth filtration and absorption. Microporous membrane filters got here into widespread use for sterilization of pharmaceutical options through the Nineteen Fifties. Use of these filters required integrity tests, such because the bubble level, which could be correlated to the size of the largest pore(s) within the membrane, paving the way in which for validation studies to demonstrate the flexibility of a membrane to remove specific microorganisms from an answer. The first microporous membranes used for sterilizing filtration had a pore dimension designation of 0. The discovery that small bacteria might exist in pharmaceutical process streams beneath conditions the place nutrient ranges were extraordinarily low led to the search for membrane filters of nonetheless smaller pore dimension. Use of these filters restricts throughput and leads to comparatively long filtration times, and 0. Nanofilters Future of Parenteral Manufacturing are sometimes used for virus removal within the pharmaceutical and biopharmaceutical industries. It is unlikely that filters with smaller pore-size ratings will be used except new technologies are found to enhance throughput and to scale back expense. The container�closure system is produced in a cleanroom, providing efficient management over visible particulate matter. Leachable compounds from the polymers, plasticizers, inks, and label adhesives can doubtlessly find their way into the completed product. Plastic containers may be extra permeable to atmospheric gases and water vapor than glass. Oxygen-sensitive merchandise require careful consideration of gas permeation properties in the growth course of; however, correct choices can present a vapor barrier corresponding to glass. Again, careful formulation development and packaging configurations using vapor-resistant barrier overwraps can mitigate water vapor and gasoline transfer issues. The share of plastic containers used for pharmaceutical packaging has been steadily increasing. It is likely to continue to enhance with the invention of new polymers and packaging technologies that will cut back vapor transmission and leachables, enhance container readability, and provide increased security for highly potent products. Industry has seen remarkable improvement within the properties of plastic containers, and we suspect there will be an much more fast rate of plastics use in delivery methods and containers in the future. Filling Plastic Containers Glass vials, ampuls, and bottles are being used more extensively to bundle parenteral products. Glass containers are clear, supply high chemical resistance, and can be securely closed either by fusion or with elastomeric closures, thereby preserving product sterility. These containers additionally face up to autoclaving and have low vapor and gasoline transmission characteristics. Failure to adequately prepare glass containers can outcome in undesirable particulate matter within the completed product. Breakage can be especially problematic if the pharmaceutical products are highly potent, poisonous, or carcinogenic. Plastic containers clear up many of those problems while offering some new ones of their very own. The first widespread use of plastic containers for injectable merchandise was for large-volume parenterals. However, particular autoclave cycles needed to be developed to prevent damage to the containers throughout post-filling terminal sterilization. There are actually infusion bags during which the drug and diluent are aseptically crammed in advanced applied sciences similar to automation and isolator know-how. Like infusion baggage, the containers are translucent, which hampers visual inspection of the contents. Closed Vial Technology Glass vials and elastomeric closures have been used for many years to package parenteral merchandise. The vials and closures should be washed and depyrogenated earlier than use and within the case of aseptically filled merchandise rendered sterile. The managed setting is monitored to guarantee it remains inside its designed operating parameters. Product contact surfaces of the vials and closures should remain sterile throughout the filling process. This implies that stopper hoppers, tracks, and vibratory bowls should be sterilized utilizing validated processes and assembled so that they remain sterile throughout the filling and stoppering processes. Closed vial filling significantly decreases energy consumption, eliminates the prices of set up and operational qualification of unneeded utility and environmental management methods, and reduces validation prices, particularly those related to environmental monitoring and media fills. The closed vial filling system incorporates inline e-beam irradiation for floor sterilization of the closure instantly previous to filling by way of the vial closure (stopper), laser resealing of the closure puncture, and application of the flip-away cap. Other advantages embody high levels of safety for operators, supply-chain, and medical personnel when dealing with potent or cytotoxic products because the process design uses polymeric vials that remove breakage. The subsequent generation of this expertise adds a closed needle which enters the closed container, opens, dispenses the liquid, and closes before exiting the container. Parenteral Medications � Elimination of aseptic gowning for personnel � Reduction in exterior environmental situations relative to other technologies � Reduction in operating costs � Superior environmental situations For these causes and others, the pattern toward elevated use of isolators is more likely to continue. Although isolators have noteworthy advantages, they will not be your greatest option for all product varieties or all drug supply system/drug product mixtures. When we think of isolation technology, we immediately consider vaporphase hydrogen peroxide decontamination of the system as nicely. However, protein and peptide merchandise can in some circumstances be extraordinarily delicate to H2O2 residues.
Discount lisinopril 2.5mg free shippingCommon excipients that readily crystallize, in addition to sodium chloride and sodium phosphate dibasic, are mannitol, glycine, and polyethylene glycols. The glassy mixture resulting from freeze focus does endure a glass transition because the temperature decreases, the place the viscosity of the mixture may enhance by orders of magnitude over a temperature vary of a few levels. The glass transition temperature of the maximally freeze-concentrated solute, known as Tg, is the physical chemical foundation for collapse in freeze-drying. If the temperature of the product is held under Tg, the glassy combination of solute and unfrozen water is inflexible enough to help its own weight as the supporting structure of ice crystals is sublimed away. This results in retention of the microstructure that was established by the freezing course of. If, nevertheless, the temperature of the system is elevated above Tg throughout major drying, the glassy combination of solute and water can bear viscous circulate under the pressure of gravity when ice is sublimed, a phenomenon generally identified as collapse. A pharmaceutically acceptable freeze-dried strong usually has the identical measurement and form as the liquid that was originally stuffed into the vials, and has a uniform color and texture. In addition, collapse ends in a lower within the particular surface space of the freeze-dried solids, and this may find yourself in longer reconstitution time relative to a system that retains the microstructure established by freezing. Perhaps extra importantly, collapsed systems are likely to have greater levels of residual moisture, maybe because of decreased floor area obtainable for evaporation of the water that was part of the glassy mixture. It is necessary to recognize that both Tg and collapse temperatures are more subjective measurements than, for instance, melting temperatures. This is mentioned additional the thermal evaluation part under coping with characterization of frozen methods. Solutions of sodium phosphate, the commonest buffer in freeze-dried formulations, are known to bear a pH shift accompanying freezing, such that the efficient pH in the freeze focus fashioned during freezing could be significantly lower than that of the beginning answer. This happens because the dibasic buffer salt is much less soluble at low temperature than the monobasic salt. Gomez and Rodriguez-Hornedo (10) used a particular pH electrode designed to face up to freezing to study the influences of preliminary buffer resolution pH and focus on subsequent pH modifications during freezing, in addition to the affect of other species in answer on buffer salt crystallization. These investigators reported that the pH modifications related to crystallization of a sodium phosphate buffer solution initially at pH 7. Further, the decrease the preliminary pH of the buffer, the upper the noticed pH at -10�C. Addition of NaCl will increase the ion product of dibasic sodium phosphate, thereby resulting in bigger pH modifications. Solutes such as sucrose and mannitol inhibited crystallization of buffer species, resulting in smaller pH shifts upon freezing. The presence of sucrose and mannitol at concentrations above three moles per mole of dibasic sodium phosphate fully prevented buffer salt crystallization. Other components of formulation, particularly people who stay amorphous throughout and after freezing, as nicely as fast freezing charges, are probably to inhibit this crystallization. Larsen (11) reported that Freeze-Drying 915 of most pharmaceutically relevant supplies tend to be fairly high-in the range of -1�C to about -15�C. Glass transition temperatures, then again, range over a much wider range (see Table forty two. In addition to different habits during freeze-drying, the physical state of the drug can dramatically influence the steadiness of the freeze-dried solid. Amorphous medicine can bear solidstate degradation at substantially larger rates than the same drug as a crystalline stable (12). In this case, the freeze-dry cycle conditions have to be based on the lowest of both the eutectic melting temperature or the collapse temperature. Annealing is simply heating the "frozen" system after the initial freezing process-not sufficient to melt the product, but sufficient to promote crystallization of parts of the formulation that have initially formed glassy mixtures. Typically, an annealing step would encompass heating the frozen system to a temperature higher than Tg but lower than the onset of melting, and holding for 2�3 h. Gatlin and DeLuca (13) investigated three cephalosporins that each one type glassy mixtures upon preliminary freezing and, until annealed, remained in the less fascinating amorphous kind after freeze-drying. There are necessary variations in freeze-drying behavior between methods the place the solute crystallizes and those the place it stays amorphous. Amorphous systems, on the opposite hand, comprise a major amount of unfrozen water. Maximally freeze-concentrated sucrose, for example, contains about 20% unfrozen water, which requires removing during secondary drying. Second, eutectic melting temperatures Eutectic Mixture Glass Solute Forms a Lyotropic Liquid Crystal States of matter which have levels of order intermediate between amorphous and crystalline are known as liquid crystals. Liquid crystals are broadly categorized as thermotropic, which are fashioned by heating, and lyotropic, that are shaped by the addition of solvent to a strong. Compounds which type liquid crystals are usually surface energetic, and the liquid crystal represents a more ordered construction than a micelle. These larger ordered structures are a results of freeze concentration, and may be either lamellar or rod formed. There have been few stories of lyotropic liquid crystal formation in aqueous solutions of drugs, and even fewer that are related to freeze-drying. Powell and coworkers (14) reported peptide liquid crystal formation by the luteinizing hormone-releasing hormone deterelix and the effect of added salts on thermodynamic stability of the liquid crystal part. Freeze-drying of frozen methods containing lyotropic mesophases appears to lead to a unique X-ray diffractogram consisting of a single sharp peak at low angle (less than about 5� 2 in addition to the "halo" which is characteristic of amorphous solids). The influence of liquid crystal formation during freezing on important high quality attributes of freeze-dried merchandise is a subject that continues to be largely unexplored. The glass transition of the maximally freeze-concentrated solute is noticed as a shift in the baseline towards higher heat capacity. First, eutectic melting might take place at a temperature very close to the melting endotherm of ice; for example, eutectic mixtures of mannitol/ice and dibasic sodium phosphate/ ice undergo eutectic melting at about -1�C and -0. Therefore, decision of eutectic melting from ice melting could be a vital source of uncertainty. Fragile glasses have comparatively slim glass transition regions, and relatively high warmth capacity change related to the glass transition. Strong glasses are the opposite-they have broad glass transition regions and small heat capability change related to this transition. Fortunately, most pharmaceutically relevant amorphous supplies (with the exception of proteins) are fragile glasses. Finally, the glass transition area, particularly for methods containing more than about 10% of an amorphous solute, could additionally be observed as more than a single transition. Sacha and coworkers (22) have proven that disaccharides share this "double Characterization of Freezing Behavior the purpose of characterizing the freezing behavior of a formulation supposed for freeze-drying is primarily to determine the maximum allowable product temperature during the main drying part, as well as to acquire insight into the bodily state of the fabric throughout and after freeze-drying. Thermal Analysis Thermal evaluation of frozen methods supposed for freeze-drying has turn into a standard device for formulation and process growth (19�21). Physical or chemical modifications in a material occurring with adjustments in temperature are accompanied by the absorption or launch of vitality within the type of warmth. In this methodology, the temperature is modified linearly with superposed sinusoidal temperature modulation, and the sample thermal response is observed in comparison with that of the thermally inert reference material. The reader is referred to evaluations by Coleman and Craig (23), Schawe (24), and Ozawa (25). It is helpful to use a microscope with polarizing functionality to have the ability to achieve information about the physical state of the pattern.
Purchase lisinopril 10 mg on-lineAdded Salts Salts such as sodium chloride are often included in freeze-dried formulations to present an isotonic reconstituted answer. First, when utilized in mixture with amorphous excipients, added salt tends to decrease the collapse temperature (discussed below), making the process less efficient and, in some instances, growing the danger of not having the power to make a pharmaceutically acceptable product (8). Added salt can also inhibit crystallization of elements of the formulation for which crystallization is needed. Bulking Agents Bulking agents, mentioned above, are needed when the drug amount is insufficient to type a pharmaceutically acceptable freeze-dried stable, and the drug is dispersed in an inert matrix that has acceptable freeze-drying traits. Bulking agents fall into two common categories-those that are likely to crystallize from a frozen system and those who stay amorphous. Polyethylene glycols, which are much less frequent, additionally are inclined to crystallize from freezing solutions. Whether these excipients actually crystallize relies upon largely on their focus relative to different formulation elements and, to a lesser extent, on the thermal history of freezing. Crystallizing excipients have the benefit of permitting freeze-drying at relatively excessive product temperatures (see discussion below), which provides more efficient processing. Mannitol is thought to , in some instances, promote vial breakage during freeze-drying (3). In addition to causing loss of yield of acceptable product, this will create issues with containment of cytotoxic compounds. Rates of breakage improve considerably when the relative fill quantity exceeds about onethird of the capability of the vial. The vial specs may play a role-particularly the heel radius, where the Freezing Process Freezing is a crucial step in the freeze-drying process, because the physical state of the frozen system influences quality attributes of the ultimate product, as properly as the process efficiency. Characterization of freezing conduct is a vital step in the development of a freeze-dried product for several reasons. First, the driving pressure for freeze-drying is the vapor stress of ice, and the vapor pressure of ice is very temperature dependent. A aim of process optimization is to carry out freeze-drying on the highest price attainable without inflicting injury to the product. Every frozen formulation has an higher temperature restrict (more about this below) in the course of the major drying course of, and it may be very important know this higher temperature limit and use it in process development so that the product temperature stays safely beneath this restrict during primary drying, however not thus far beneath the limit as to make the process unnecessarily time consuming. Second, process validation entails assuring that the cycle conditions are acceptable for the formulation. In order to be ready to validate a process, "benchmark" information must be out there to assess the adequacy of the process situations. Regulatory authorities expect a scientific rationale for freeze-dry cycle conditions, with appropriate documentation. Parenteral Medications which in flip influences ice crystal morphology and the amount of water in the system that remains unfrozen. It is important to first think about ice morphology before contemplating the behavior of elements of a formulation during freezing. Different ice morphologies, together with regular and irregular dendrites, in addition to spherulitic systems (thin fibers if ice radiating outward from the nucleation site), type throughout freezing, relying on the freezing fee and the sort and concentration of solutes current. Ice crystal morphology and measurement distribution have since been shown to influence the charges of main (9) and secondary drying, in addition to protein aggregation in freeze-dried protein formulations. This is adopted by secondary nucleation, during which a visible entrance propagates by way of some portion of the pattern at a price on the order of a number of millimeters per second. This process stops as the temperature of the system approaches the equilibrium freezing temperature. Secondary nucleation is followed by solidification, which takes place at a slower rate as the heat released by ice crystallization is conducted out of the sample and ultimately to the heat switch fluid. In the primary, termed global supercooling, the complete liquid volume reaches the same degree of supercooling, and the secondary nucleation zone contains the whole answer volume. In directional solidification, a portion of the liquid quantity is cooled to the point of major and secondary nucleation, and the nucleation and solidification fronts move together into the beforehand unnucleated portion of the solution. Supercooling is essential as a result of the actual freezing rate of an answer is set by the time elapsed between nucleation of the first ice crystals and complete solidification of the system. This is usually confused with the cooling rate, which is the rate at which the temperature of the shelves is decreased during freezing. Ice nucleation, like all crystallization course of, can be either homogeneous, the place water molecules spontaneously order themselves into nuclei, or heterogeneous, the place nucleation is triggered by a surface or by extraneous particulate matter. Nevertheless, there are still irregularities within the microstructure of the glass that may function nucleation sites, but aqueous options intended to be freezedried can supercool by as a lot as 12�C�15�C before ice crystals nucleate. However, directional solidification usually requires some sort of ice nucleating agent. The freezing mechanism was demonstrated to be mirrored within the morphology of the freeze-dried cake. If an answer of regular saline is frozen, for instance, the sodium chloride concentration within the preliminary solution is 0. When this solution is frozen, the sodium chloride concentration in the interstitial house between ice crystals reaches almost four N earlier than sodium chloride co-crystallizes as a eutectic combination. In the case of formulations containing sodium chloride or other salts, this high ionic power environment can be damaging, significantly to biological materials corresponding to proteins and cells. After ice crystal development has essentially completed, and the solute has been concentrated as much as possible within the interstitial house between ice crystals, what happens subsequent is dependent upon whether or not the solute crystallizes from this freeze focus. If the system is cooled to a temperature under point C, the solute is now not soluble, and it crystallizes and precipitates. This is the eutectic (from Greek, that means "easily melted") composition, and the temperature corresponding to C is the eutectic melting temperature. What the part diagram tells us about freeze-drying of a solution of sodium chloride in water is the next. In the twophase region, because the temperature is decreased, the ice crystals grow and the freeze focus turns into more concentrated. At any temperature on this area, the composition of the system is given by a horizontal line (called a "tie line") via this temperature. When this concentration reaches level C, a eutectic mixture of sodium chloride and ice precipitates from the freeze concentrate. In actuality, the crystallization of solute is simply as unpredictable because the crystallization of water. Eutectic mixtures melt at a sharply defined temperature, as if they have been a single, pure compound. The significance of the eutectic melting temperature to freeze-drying is that it represents the maximum allowable product temperature throughout main drying. Exceeding this temperature during the course of would end in puffing, foaming, perhaps expulsion of solids from a vial, and loss of pharmaceutical acceptability.
References - Goldman HB, Rackley RR, Appell RA: The efficacy of urethrolysis without re-suspension for iatrogenic urethral obstruction, J Urol 161:1268n1271, 1999.
- Torikai S, Kurokawa K: Effect of PGE2 on vasopressin-dependent cell cAMP in isolated single nephron segments, Am J Physiol 245(1):F58nF66, 1983.
- Tyritzis SI, Hosseini A, Jonsson M, et al: Robot-assisted intracorporeal formation of the ileal neobladder, J Endourol 26:1570n1575, 2012.
|
|


